 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1j9w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Solution Structure of the CAI Michigan 1 Variant | ||||||
 Components Components | CARBONIC ANHYDRASE I Carbonic anhydrase Carbonic anhydrase | ||||||
 Keywords Keywords | LYASE / 10-stranded-beta-sheet | ||||||
| Function / homology |  Function and homology informationhydro-lyase activity / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / Reversible hydration of carbon dioxide / carbonic anhydrase / carbonate dehydratase activity / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen ...hydro-lyase activity / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / Reversible hydration of carbon dioxide / carbonic anhydrase / carbonate dehydratase activity / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / one-carbon metabolic process / extracellular exosome / zinc ion binding / cytosol Function and homology informationhydro-lyase activity / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / Reversible hydration of carbon dioxide / carbonic anhydrase / carbonate dehydratase activity / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen ...hydro-lyase activity / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / Reversible hydration of carbon dioxide / carbonic anhydrase / carbonate dehydratase activity / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / one-carbon metabolic process / extracellular exosome / zinc ion binding / cytosolSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Briganti, F. / Ferraroni, M. / Chedwiggen, W.R. / Scozzafava, A. / Supuran, C.T. / Tilli, S. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: Crystal structure of a zinc-activated variant of human carbonic anhydrase I, CA I Michigan 1: evidence for a second zinc binding site involving arginine coordination. Authors: Ferraroni, M. / Tilli, S. / Briganti, F. / Chegwidden, W.R. / Supuran, C.T. / Wiebauer, K.E. / Tashian, R.E. / Scozzafava, A. #1: Journal: Science / Year: 1962 Title: New genetically determined molecular form of erythrocyte esterase in man. Authors: SHAW, C.R. / SYNER, F.N. / TASHIAN, R.E. #2: Journal: Hum.Mutat. / Year: 1994 Title: Marked zinc activation of ester hydrolysis by a mutation, 67-His (CAT) to Arg (CGT), in the active site of human carbonic anhydrase I. Authors: Chegwidden, W.R. / Wagner, L.E. / Venta, P.J. / Bergenhem, N.C. / Yu, Y.S. / Tashian, R.E. #3: Journal: GENE FAM.LSOZYME BULL. / Year: 1998Title: Point Mutation in the Variant of Human Carbonic Anhydrase Isozyme I (CAI Michigan ) Active Site decreases affinity for aromatic sulfonamide inhibitors Authors: Briganti, F. / Chegwidden, W.R. / Scozzafava, A. / Supuran, C.T. / Tashian, R.E. / Wiebauer, K.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1j9w.cif.gz | 120.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1j9w.ent.gz | 91.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1j9w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j9/1j9wftp://data.pdbj.org/pub/pdb/validation_reports/j9/1j9w | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 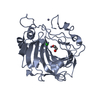 1jv0C  1czmS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | the biological assembly is a dimer generated by a translation |
-Components
| #1: Protein | Carbonic anhydrase / CARBONATE DEHYDRATASE I Mass: 28794.035 Da / Num. of mol.: 2 / Mutation: H67R Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: pKK232-2 / Production host:  Escherichia coli (E. coli) / Strain (production host): JM109 / References: UniProt: P00915, carbonic anhydrase Escherichia coli (E. coli) / Strain (production host): JM109 / References: UniProt: P00915, carbonic anhydrase#2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | Ethylene glycol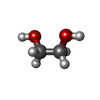  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 321 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 321 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.25 Å3/Da / Density % sol: 45 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 9 Details: PEG 4000, litium chloride,ethylene glicol , pH 9.0, VAPOR DIFFUSION, HANGING DROP, temperature 295K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: MACSCIENCE M12X / Wavelength: 1.5418 Å |
| Detector | Type: BRUKER SMART 1000 / Detector: CCD / Date: May 26, 2000 / Details: mirrors |
| Radiation | Monochromator: Gobel mirrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→20 Å / Num. all: 58715 / Num. obs: 58715 / % possible obs: 97.9 % / Redundancy: 3.3 % / Biso Wilson estimate: 24.36 Å2 / Rsym value: 0.094 / Net I/σ(I): 7.8 |
| Reflection shell | Resolution: 2.6→2.69 Å / Mean I/σ(I) obs: 1.5 / Num. unique all: 17135 / Rsym value: 0.244 / % possible all: 100 |
| Reflection | *PLUS Lowest resolution: 20 Å / Num. obs: 17329 / Num. measured all: 58715 / Rmerge(I) obs: 0.094 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdb entry 1czm Resolution: 2.6→15 Å Isotropic thermal model: isotropic, anisotropic for metal ions, sulfur atoms, choride ions Cross valid method: THROUGHOUT / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.82 Å2 | ||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.6 Å / Lowest resolution: 15 Å / % reflection Rfree: 5 % / Rfactor obs: 0.197 | ||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
