Mass: 18.015 Da / Num. of mol.: 264 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has ligand of interest
Y
Sequence details
Chains A and C, as well as B and D, were manipulated as one single peptide, but auto-cleaved at ...Chains A and C, as well as B and D, were manipulated as one single peptide, but auto-cleaved at residue 256 during zymogen activation.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.45 Å3/Da / Density % sol: 49.69 %
Crystal grow
Temperature: 293 K / Method: vapor diffusion, hanging drop Details: 0.1M acetic acid/sodium acetate, pH 4.0, 20% w/v PEG 4000
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords HYDROLASE / Inhibitor /
HYDROLASE / Inhibitor /  Function and homology information
Function and homology information
 Authors
Authors China, 1items
China, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
 PDBj
PDBj


 Assembly
Assembly


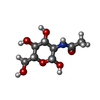
 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H15NO6 / Feature type: SUBJECT OF INVESTIGATION
Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H15NO6 / Feature type: SUBJECT OF INVESTIGATION


 Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca / Feature type: SUBJECT OF INVESTIGATION
Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca / Feature type: SUBJECT OF INVESTIGATION Mass: 179.176 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H9N3O2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 179.176 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H9N3O2 / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation Processing
Processing