 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2hmw: Square-shaped octameric ring structure of an RCK domain with ATP bound -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2hmw | ||||||
|---|---|---|---|---|---|---|---|
| Title | Square-shaped octameric ring structure of an RCK domain with ATP bound | ||||||
 Components Components | YuaA protein Yuya Yuya | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / RCK / KTN / KTR / KTRA / KTRAB / membrane protein / ion transporter / symporter | ||||||
| Function / homology |  Function and homology information Function and homology informationmonoatomic cation transmembrane transporter activity / potassium ion transport / identical protein binding / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Bacillus subtilis (bacteria) Bacillus subtilis (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å | ||||||
 Authors Authors | Albright, R.A. / Morais-Cabral, J.H. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2006 Title: The RCK Domain of the KtrAB K(+) Transporter: Multiple Conformations of an Octameric Ring. Authors: Albright, R.A. / Ibar, J.L. / Kim, C.U. / Gruner, S.M. / Morais-Cabral, J.H. #1: Journal: Cell(Cambridge,Mass.) / Year: 2002Title: A Mechanism of Regulating Transmembrane Potassium Flux through a Ligand-Mediated Conformational Switch Authors: Roosild, T.P. / Miller, S. / Booth, I.R. / Choe, S. #2: Journal: Nature / Year: 2002Title: Crystal structure and mechanism of a calcium-gated potassium channel Authors: Jiang, Y. / Lee, A. / Chen, J. / Cadene, M. / Chait, B.T. / MacKinnon, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2hmw.cif.gz | 67.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2hmw.ent.gz | 50.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2hmw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hm/2hmwftp://data.pdbj.org/pub/pdb/validation_reports/hm/2hmw | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2hmsC  2hmtC  2hmuC  2hmvC  1lsuS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is an octameric ring generated from the asymmetric unit dimer using these symmetry operators: X, Y, Z and Y, 1-X, Z and 1-X, 1-Y, Z and 1-Y, X, Z |
-Components
| #1: Protein | Yuya Mass: 16076.447 Da / Num. of mol.: 2 / Fragment: RCK CORE DOMAIN (KTN), residues 1-144 / Mutation: C22V Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacillus subtilis (bacteria) / Gene: yuaA / Plasmid: pET15b / Species (production host): Escherichia coli / Production host: Escherichia coli BL21(DE3) (bacteria) / Strain (production host): BL21(DE3) / References: UniProt: O32080#2: Chemical | Adenosine triphosphate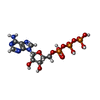  Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.06 Å3/Da / Density % sol: 59.76 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 8.8 Details: 500mM MgCl2, 50mM Tris pH 8.8, 8.5% (w/v) PEG 2000, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1.1 Å / Beamline: X25 / Wavelength: 1.1 Å |
|---|---|
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Sep 15, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 3→50 Å / Num. all: 8258 / Num. obs: 8231 / % possible obs: 99.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 9.9 % / Rmerge(I) obs: 0.1 / Χ2: 1.007 / Net I/σ(I): 11.1 |
| Reflection shell | Resolution: 3→3.11 Å / Redundancy: 10.2 % / Rmerge(I) obs: 0.731 / Num. unique all: 799 / Χ2: 1.007 / % possible all: 99.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: single domain from pdb entry 1LSU Resolution: 3→50 Å / FOM work R set: 0.756 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 50.849 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 84.146 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 17
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
