 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1do0: ORTHORHOMBIC CRYSTAL FORM OF HEAT SHOCK LOCUS U (HSLU) FROM ESCHE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1do0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | ORTHORHOMBIC CRYSTAL FORM OF HEAT SHOCK LOCUS U (HSLU) FROM ESCHERICHIA COLI | ||||||
 Components Components | PROTEIN (HEAT SHOCK LOCUS U) | ||||||
 Keywords Keywords | CHAPERONE / HSLU / CLPY / AAA-ATPASE / ATP-DEPENDENT PROTEOLYSIS / ATPASE / PROTEASOME | ||||||
| Function / homology |  Function and homology information Function and homology informationprotein denaturation / HslUV protease complex / proteasome-activating activity / protein unfolding / : / peptidase activity / cellular response to heat / response to heat / protein domain specific binding / magnesium ion binding ...protein denaturation / HslUV protease complex / proteasome-activating activity / protein unfolding / : / peptidase activity / cellular response to heat / response to heat / protein domain specific binding / magnesium ion binding / ATP hydrolysis activity / proteolysis / ATP binding / identical protein binding / membrane / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3 Å | ||||||
 Authors Authors | Bochtler, M. / Hartmann, C. / Song, H.K. / Bourenkov, G.P. / Bartunik, H.D. | ||||||
 Citation Citation | Journal: Nature / Year: 2000 Title: The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Authors: Bochtler, M. / Hartmann, C. / Song, H.K. / Bourenkov, G.P. / Bartunik, H.D. / Huber, R. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1997Title: Crystal Structure of Heat Shock Locus V (HslV) from Escherichia coli Authors: Bochtler, M. / Ditzel, L. / Groll, M. / Huber, R. #2: Journal: Proc.Natl.Acad.Sci.USA / Year: 1996Title: HslV-HslU: A Novel ATP-Dependent Protease Complex in Escherichia coli Related to the Eukaryotic Proteasome Authors: Rohrwild, M. / Coux, O. / Huang, H.C. / Moerschell, R.P. / Yoo, S.J. / Seol, J.H. / Chung, C.H. / Goldberg, A.L. #3: Journal: Gene / Year: 1993Title: Sequence Analysis of Four New Heat-Shock Genes Constituting the hslTS/ibpAB and hslVU Operons in Escherichia coli Authors: Chuang, S.E. / Burland, V. / Plankett III, G. / Daniels, D.L. / Blattner, F.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1do0.cif.gz | 480.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1do0.ent.gz | 391.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1do0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/do/1do0ftp://data.pdbj.org/pub/pdb/validation_reports/do/1do0 | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 49528.508 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical | ChemComp-ATP / 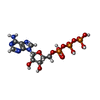  Mass: 507.181 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM#3: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4Sequence details | EDMAN-DEGRADATION HAS SHOWN THAT THE AMINOTERMINAL METHIONINE IS CLEAVED IN THE WILD TYPE. ...EDMAN-DEGRADATIO | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.18 Å3/Da / Density % sol: 61.27 % | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: reservoir: 100 mM sodium cacodylate, 15% glycerol, 10.5% PEG8000, 500 mM (NH4)2(SO4). drop: 0.002 ml protein (16mg/ml) in 20mM Tris/HCl pH 7.5, 1mM EDTA, 1mM NaN3, 0.002 ml reservoir, 0.0005 ...Details: reservoir: 100 mM sodium cacodylate, 15% glycerol, 10.5% PEG8000, 500 mM (NH4)2(SO4). drop: 0.002 ml protein (16mg/ml) in 20mM Tris/HCl pH 7.5, 1mM EDTA, 1mM NaN3, 0.002 ml reservoir, 0.0005 ml 1.0 M guanidinium chloride, 0.0005 ml C12E8 (octaethylene glycol monododecyl ether), pH 6.5, VAPOR DIFFUSION, SITTING DROP, temperature 291K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Details: drop consists of equal volume of protein and reservoir solutions in Buffer B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1.07 / Beamline: BW6 / Wavelength: 1.07 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 15, 1999 / Details: MIRROR |
| Radiation | Monochromator: DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.07 Å / Relative weight: 1 |
| Reflection | Resolution: 3→35 Å / Num. all: 265668 / Num. obs: 74040 / % possible obs: 96.9 % / Redundancy: 3 % / Biso Wilson estimate: 55.1 Å2 / Rmerge(I) obs: 0.079 / Rsym value: 7.9 |
| Reflection shell | Resolution: 3→3.05 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.526 / Rsym value: 52.6 / % possible all: 96 |
| Reflection | *PLUS Num. measured all: 265668 |
| Reflection shell | *PLUS % possible obs: 96 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: HSLU FROM HSLU IN HSLVU CRYSTALS Resolution: 3→25 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 2596651.04 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 54.86 Å2 / ksol: 0.254 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 55.1 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3→3.19 Å / Rfactor Rfree error: 0.018 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.9 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
