 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ae5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN HEPARIN BINDING PROTEIN | ||||||
 Components Components | HEPARIN BINDING PROTEIN | ||||||
 Keywords Keywords | SERINE PROTEASE HOMOLOG /  ENDOTOXIN BINDING / HEPARIN BINDING / INFLAMMATION / ANTIBACTERIAL / CAP37 / AZUROCIDIN / GLYCOSYLATED PROTEIN ENDOTOXIN BINDING / HEPARIN BINDING / INFLAMMATION / ANTIBACTERIAL / CAP37 / AZUROCIDIN / GLYCOSYLATED PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationneutrophil-mediated killing of bacterium / monocyte activation / cellular extravasation / positive regulation of fractalkine production / protein kinase C signaling / glial cell migration / regulation of vascular permeability / induction of positive chemotaxis / protein kinase C-activating G protein-coupled receptor signaling pathway / positive regulation of MHC class II biosynthetic process ...neutrophil-mediated killing of bacterium / monocyte activation / cellular extravasation / positive regulation of fractalkine production / protein kinase C signaling / glial cell migration / regulation of vascular permeability / induction of positive chemotaxis / protein kinase C-activating G protein-coupled receptor signaling pathway / positive regulation of MHC class II biosynthetic process / antimicrobial humoral response / heparan sulfate proteoglycan binding / azurophil granule / azurophil granule membrane / macrophage chemotaxis / : / positive regulation of cell adhesion / toxic substance binding / positive regulation of protein kinase activity / positive regulation of phagocytosis / cell chemotaxis / positive regulation of peptidyl-threonine phosphorylation / positive regulation of interleukin-1 beta production / microglial cell activation / azurophil granule lumen / positive regulation of tumor necrosis factor production / heparin binding / peptidase activity / defense response to virus / defense response to Gram-negative bacterium / inflammatory response / serine-type endopeptidase activity / intracellular membrane-bounded organelle / Neutrophil degranulation / positive regulation of gene expression / negative regulation of apoptotic process / proteolysis / extracellular space / extracellular exosome / extracellular region / membraneSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Iversen, L.F. / Kastrup, J.S. / Bjorn, S.E. / Rasmussen, P.B. / Wiberg, F.C. / Flodgaard, H.J. / Larsen, I.K. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1997 Title: Structure of HBP, a multifunctional protein with a serine proteinase fold. Authors: Iversen, L.F. / Kastrup, J.S. / Bjorn, S.E. / Rasmussen, P.B. / Wiberg, F.C. / Flodgaard, H.J. / Larsen, I.K. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1996Title: Crystallization and Molecular Replacement Solution of Human Heparin Binding Protein Authors: Iversen, L.F. / Kastrup, J.S. / Larsen, I.K. / Bjorn, S.E. / Rasmussen, P.B. / Wiberg, F.C. / Flodgaard, H.J. #2: Journal: FEBS Lett. / Year: 1996Title: Characterization of Recombinant Human Hbp/CAP37/Azurocidin, a Pleiotropic Mediator of Inflammation-Enhancing Lps-Induced Cytokine Release from Monocytes Authors: Rasmussen, P.B. / Bjorn, S. / Hastrup, S. / Nielsen, P.F. / Norris, K. / Thim, L. / Wiberg, F.C. / Flodgaard, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ae5.cif.gz | 53.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ae5.ent.gz | 40.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1ae5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ae/1ae5ftp://data.pdbj.org/pub/pdb/validation_reports/ae/1ae5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1ppfS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24302.475 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell line: SF-900 / Cell line (production host): SF-900 / Production host:  unidentified baculovirus / Strain (production host): ACMNPV / References: UniProt: P20160 unidentified baculovirus / Strain (production host): ACMNPV / References: UniProt: P20160 | ||
|---|---|---|---|
| #2: Sugar | N-Acetylglucosamine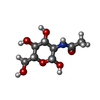  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.2 / Details: pH 7.2 | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: Iversen, L.F., (1996) Acta Crystallogr.,Sect.D, 52, 1222. | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 300 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.999 / Beamline: X11 / Wavelength: 0.999 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Sep 1, 1996 / Details: MIRRORS |
| Radiation | Monochromator: MIRRORS / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.999 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→20 Å / Num. obs: 12176 / % possible obs: 99.2 % / Observed criterion σ(I): 1 / Redundancy: 4.7 % / Rmerge(I) obs: 0.089 / Rsym value: 0.089 / Net I/σ(I): 14.3 |
| Reflection shell | Resolution: 2.3→2.34 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.264 / Mean I/σ(I) obs: 4.9 / Rsym value: 0.264 / % possible all: 98.6 |
| Reflection shell | *PLUS % possible obs: 98.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1PPF Resolution: 2.3→20 Å / σ(F): 1 Details: THERE IS A DISORDERED REGION IN THE STRUCTURE INVOLVING RESIDUES 44 - 48.
| ||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→20 Å
| ||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | ||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor all: 0.192 / Rfactor Rfree: 0.266 / Rfactor Rwork: 0.192 | ||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
