 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8x2r | ||||||
|---|---|---|---|---|---|---|---|
| Title | The Crystal Structure of HSP 90-alpha from Biortus. | ||||||
 Components Components | Heat shock protein HSP 90-alpha Heat shock response Heat shock response | ||||||
 Keywords Keywords | CHAPERONE / Host-virus interaction / ATP-binding | ||||||
| Function / homology |  Function and homology information Function and homology informationsperm mitochondrial sheath / dATP binding / Scavenging by Class F Receptors / sulfonylurea receptor binding / CTP binding / positive regulation of protein polymerization / vRNP Assembly / UTP binding / sperm plasma membrane / positive regulation of tau-protein kinase activity ...sperm mitochondrial sheath / dATP binding / Scavenging by Class F Receptors / sulfonylurea receptor binding / CTP binding / positive regulation of protein polymerization / vRNP Assembly / UTP binding / sperm plasma membrane / positive regulation of tau-protein kinase activity / protein insertion into mitochondrial outer membrane / chaperone-mediated autophagy / telomerase holoenzyme complex assembly / Rho GDP-dissociation inhibitor binding / Uptake and function of diphtheria toxin / mitochondrial transport / Drug-mediated inhibition of ERBB2 signaling / Resistance of ERBB2 KD mutants to trastuzumab / Resistance of ERBB2 KD mutants to sapitinib / Resistance of ERBB2 KD mutants to tesevatinib / Resistance of ERBB2 KD mutants to neratinib / Resistance of ERBB2 KD mutants to osimertinib / Resistance of ERBB2 KD mutants to afatinib / Resistance of ERBB2 KD mutants to AEE788 / Resistance of ERBB2 KD mutants to lapatinib / Drug resistance in ERBB2 TMD/JMD mutants / PIWI-interacting RNA (piRNA) biogenesis / TPR domain binding / non-chaperonin molecular chaperone ATPase / regulation of postsynaptic membrane neurotransmitter receptor levels / dendritic growth cone / Sema3A PAK dependent Axon repulsion / regulation of protein ubiquitination / skeletal muscle contraction / positive regulation of cell size / HSF1-dependent transactivation / telomere maintenance via telomerase / response to unfolded protein / protein unfolding / chaperone-mediated protein complex assembly / HSF1 activation / regulation of protein-containing complex assembly / Attenuation phase / RHOBTB2 GTPase cycle / positive regulation of lamellipodium assembly / eNOS activation / axonal growth cone / DNA polymerase binding / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / positive regulation of cardiac muscle contraction / Loss of Nlp from mitotic centrosomes / Loss of proteins required for interphase microtubule organization from the centrosome / Signaling by ERBB2 / Recruitment of mitotic centrosome proteins and complexes / cardiac muscle cell apoptotic process / positive regulation of telomerase activity / positive regulation of defense response to virus by host / response to salt stress / endocytic vesicle lumen / Recruitment of NuMA to mitotic centrosomes / HSP90 chaperone cycle for steroid hormone receptors (SHR) in the presence of ligand / Anchoring of the basal body to the plasma membrane / protein tyrosine kinase binding / activation of innate immune response / response to cold / positive regulation of interferon-beta production / nitric-oxide synthase regulator activity / lysosomal lumen / Constitutive Signaling by Overexpressed ERBB2 / ESR-mediated signaling / AURKA Activation by TPX2 / VEGFR2 mediated vascular permeability / response to cocaine / brush border membrane / ATP-dependent protein folding chaperone / Signaling by ERBB2 TMD/JMD mutants / neuron migration / Constitutive Signaling by EGFRvIII / DDX58/IFIH1-mediated induction of interferon-alpha/beta / Signaling by ERBB2 ECD mutants / tau protein binding / Signaling by ERBB2 KD Mutants / Regulation of necroptotic cell death / Regulation of actin dynamics for phagocytic cup formation / Downregulation of ERBB2 signaling / cellular response to virus / VEGFA-VEGFR2 Pathway / histone deacetylase binding / Aggrephagy / Chaperone Mediated Autophagy / positive regulation of protein import into nucleus / response to estrogen / positive regulation of protein catabolic process / The role of GTSE1 in G2/M progression after G2 checkpoint / regulation of protein localization / positive regulation of nitric oxide biosynthetic process / disordered domain specific binding / Regulation of PLK1 Activity at G2/M Transition / unfolded protein binding / melanosomeSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Wang, F. / Cheng, W. / Lv, Z. / Meng, Q. / Lu, Y. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: The Crystal Structure of HSP 90-alpha from Biortus. Authors: Wang, F. / Cheng, W. / Lv, Z. / Meng, Q. / Lu, Y. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8x2r.cif.gz | 110.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8x2r.ent.gz | 80.4 KB | Display | PDB format |
| PDBx/mmJSON format | 8x2r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/x2/8x2rftp://data.pdbj.org/pub/pdb/validation_reports/x2/8x2r | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|
-Links
 PDBj
PDBj



















- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Heat shock response Mass: 26585.773 Da / Num. of mol.: 1 / Mutation: S52A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HSP90AA1 / Production host:  Escherichia coli (E. coli) / References: UniProt: P07900 Escherichia coli (E. coli) / References: UniProt: P07900 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-GOL / Glycerol  Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | Ethylene glycol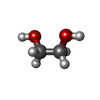  Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 62.068 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6O2 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 333 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 333 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.74 Å3/Da / Density % sol: 55.03 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / Details: (Index-H6) 0.2M Sodium formate, 20% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CLSI  / Beamline: 08ID-1 / Wavelength: 0.953698 Å / Beamline: 08ID-1 / Wavelength: 0.953698 Å |
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Aug 16, 2022 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.953698 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→36.28 Å / Num. obs: 51928 / % possible obs: 100 % / Redundancy: 8.8 % / Rmerge(I) obs: 0.061 / Net I/σ(I): 17.3 |
| Reflection shell | Resolution: 1.45→1.47 Å / Rmerge(I) obs: 0.871 / Mean I/σ(I) obs: 2.3 / Num. unique obs: 3646 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.45→34.511 Å / Cor.coef. Fo:Fc: 0.982 / Cor.coef. Fo:Fc free: 0.976 / SU B: 2.213 / SU ML: 0.036 / Cross valid method: FREE R-VALUE / ESU R: 0.048 / ESU R Free: 0.047 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.439 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→34.511 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
