SUMO fused Trehalose Synthase (TreS) of Mycobacterium tuberculosis
Components
SUMO fused Trehalose Synthase (TreS),Trehalose synthase/amylase TreS
Keywords
SUGAR BINDING PROTEIN / TreS / Trehalose Synthase / Mycobacterium tuberculosis / CYTOSOLIC PROTEIN
Function / homology
Function and homology information
maltose alpha-D-glucosyltransferase / maltose alpha-D-glucosyltransferase activity / SUMO is conjugated to E1 (UBA2:SAE1) / SUMOylation of nuclear envelope proteins / SUMO is transferred from E1 to E2 (UBE2I, UBC9) / SUMOylation of SUMOylation proteins / SUMO is proteolytically processed / SUMOylation of transcription factors / capsule polysaccharide biosynthetic process / Postmitotic nuclear pore complex (NPC) reformation ...maltose alpha-D-glucosyltransferase / maltose alpha-D-glucosyltransferase activity / SUMO is conjugated to E1 (UBA2:SAE1) / SUMOylation of nuclear envelope proteins / SUMO is transferred from E1 to E2 (UBE2I, UBC9) / SUMOylation of SUMOylation proteins / SUMO is proteolytically processed / SUMOylation of transcription factors / capsule polysaccharide biosynthetic process / Postmitotic nuclear pore complex (NPC) reformation / SUMOylation of transcription cofactors / septin ring / SUMOylation of DNA damage response and repair proteins / SUMOylation of RNA binding proteins / SUMOylation of DNA replication proteins / SUMOylation of chromatin organization proteins / alpha-amylase / glycogen biosynthetic process / alpha-amylase activity / ubiquitin-like protein ligase binding / protein sumoylation / polysaccharide catabolic process / condensed nuclear chromosome / protein tag activity / identical protein binding / metal ion binding / nucleus Similarity search - Function
National Institutes of Health/National Institute Of Allergy and Infectious Diseases (NIH/NIAID)
AI105084
United States
Citation
Journal: ACS Infect Dis / Year: 2024 Title: Targeting Persistence through Inhibition of the Trehalose Catalytic Shift. Authors: Karishma Kalera / Rachel Liu / Juhyeon Lim / Rasangi Pathirage / Daniel H Swanson / Ulysses G Johnson / Alicyn I Stothard / Jae Jin Lee / Anne W Poston / Peter J Woodruff / Donald R Ronning ...Authors: Karishma Kalera / Rachel Liu / Juhyeon Lim / Rasangi Pathirage / Daniel H Swanson / Ulysses G Johnson / Alicyn I Stothard / Jae Jin Lee / Anne W Poston / Peter J Woodruff / Donald R Ronning / Hyungjin Eoh / Benjamin M Swarts / Abstract: Tuberculosis (TB), caused by (Mtb), is the leading cause of death worldwide by infectious disease. Treatment of Mtb infection requires a six-month course of multiple antibiotics, an extremely ...Tuberculosis (TB), caused by (Mtb), is the leading cause of death worldwide by infectious disease. Treatment of Mtb infection requires a six-month course of multiple antibiotics, an extremely challenging regimen necessitated by Mtb's ability to form drug-tolerant persister cells. Mtb persister formation is dependent on the trehalose catalytic shift, a stress-responsive metabolic remodeling mechanism in which the disaccharide trehalose is liberated from cell surface glycolipids and repurposed as an internal carbon source to meet energy and redox demands. Here, using a biofilm-persister model, metabolomics, and cryo-electron microscopy (EM), we found that azidodeoxy- and aminodeoxy-d-trehalose analogues block the Mtb trehalose catalytic shift through inhibition of trehalose synthase TreS (Rv0126), which catalyzes the isomerization of trehalose to maltose. Out of a focused eight-member compound panel constructed by chemoenzymatic synthesis, the natural product 2-trehalosamine exhibited the highest potency and significantly potentiated first- and second-line TB drugs in broth culture and macrophage infection assays. We also report the first structure of TreS bound to a substrate analogue inhibitor, obtained via cryo-EM, which revealed conformational changes likely essential for catalysis and inhibitor binding that can potentially be exploited for future therapeutic development. Our results demonstrate that inhibition of the trehalose catalytic shift is a viable strategy to target Mtb persisters and advance trehalose analogues as tools and potential adjunctive therapeutics for investigating and targeting mycobacterial persistence.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Mycobacterium tuberculosis /
Mycobacterium tuberculosis /  Function and homology information
Function and homology information

 Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads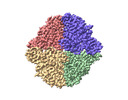

 PDBj
PDBj



 Assembly
Assembly

 Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Ca Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 7 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing