 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8sjy: Structure of lens aquaporin-0 array in sphingomyelin/cholesterol ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8sjy | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of lens aquaporin-0 array in sphingomyelin/cholesterol bilayer (1SM:2Chol) | ||||||
 Components Components | Lens fiber major intrinsic protein | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / aquaporin / lens / cholesterol / lipid raft | ||||||
| Function / homology |  Function and homology information Function and homology informationgap junction-mediated intercellular transport / water channel activity / water transport / structural constituent of eye lens / gap junction / lens development in camera-type eye / response to stimulus / positive regulation of cell adhesion / visual perception / protein homotetramerization ...gap junction-mediated intercellular transport / water channel activity / water transport / structural constituent of eye lens / gap junction / lens development in camera-type eye / response to stimulus / positive regulation of cell adhesion / visual perception / protein homotetramerization / calmodulin binding / endoplasmic reticulum / plasma membraneSimilarity search - Function | ||||||
| Biological species |  Ovis aries (sheep) Ovis aries (sheep) | ||||||
| Method | ELECTRON CRYSTALLOGRAPHY / electron crystallography / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Chiu, P.-L. / Walz, T. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Structure of aquaporin-0 arrays in sphingomyelin/cholesterol membranes and implications for lipid rafts Authors: Chiu, P.-L. / Walz, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8sjy.cif.gz | 78.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8sjy.ent.gz | 47.4 KB | Display | PDB format |
| PDBx/mmJSON format | 8sjy.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sj/8sjyftp://data.pdbj.org/pub/pdb/validation_reports/sj/8sjy | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|
-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | / Aquaporin-0 Mass: 28284.885 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Ovis aries (sheep) / Organ: Eye / Tissue: Lens / References: UniProt: Q6J8I9 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-HWP / [(   Mass: 703.028 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C39H79N2O6P / Feature type: SUBJECT OF INVESTIGATION Mass: 703.028 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C39H79N2O6P / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-CLR / Cholesterol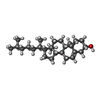  Mass: 386.654 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H46O / Feature type: SUBJECT OF INVESTIGATION Mass: 386.654 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H46O / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 11 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 11 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON CRYSTALLOGRAPHY | |||
|---|---|---|---|---|
| EM experiment | Aggregation state: 2D ARRAY / 3D reconstruction method: electron crystallography | |||
| Crystal symmetry | Image processing-ID: 1 / ∠γ: 90 ° / C sampling length: 200 Å / A: 65.5 Å / B: 65.5 Å / C: 200 Å / Space group name H-M: P422
|
- Sample preparation
Sample preparation
| Component | Name: Lens aquaporin-0 in sphingomyelin/cholesterol bilayer / Type: COMPLEX / Entity ID: #1 / Source: NATURAL | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.0283 MDa / Experimental value: NO | |||||||||||||||||||||||||
| Source (natural) | Organism: Ovis aries (sheep) / Organ: Eye / Tissue: Lens | |||||||||||||||||||||||||
| EM crystal formation | Lipid mixture: Sphingomyelin and cholesterol were mixed at a 1:2 molar ratio. Lipid protein ratio: 0.2 / Temperature: 310 K / Time: 7 DAY | |||||||||||||||||||||||||
| Buffer solution | pH: 6 Details: 10 mM MES (pH 6.0), 300 mM NaCl, 30 mM MgCl2, and 0.05% NaN3 | |||||||||||||||||||||||||
| Buffer component |
| |||||||||||||||||||||||||
| Specimen | Embedding applied: YES / Shadowing applied: NO / Staining applied: NO / Vitrification applied: NO / Details: 2D crystal of lens AQP0. | |||||||||||||||||||||||||
| Specimen support | Grid material: MOLYBDENUM / Grid type: Homemade | |||||||||||||||||||||||||
| EM embedding | Details: Aquaporin-0 2D crystals were prepared on molybdenum grids using the carbon sandwich method and a trehalose concentration ranging from 3% to 5% (w/v). Material: Trehalose |
-Data collection
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI POLARA 300 Details: The diffraction patterns were recorded without setting defocus. |
| Electron gun | Electron source: FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: DIFFRACTION / Nominal defocus max: 0 nm / Nominal defocus min: 0 nm / Calibrated defocus min: 0 nm / Calibrated defocus max: 0 nm / C2 aperture diameter: 30 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: OTHER |
| Image recording | Average exposure time: 30 sec. / Electron dose: 10 e/Å2 / Film or detector model: GATAN ULTRASCAN 4000 (4k x 4k) / Num. of diffraction images: 241 |
| EM diffraction shell | Resolution: 2.3→11.8 Å / Fourier space coverage: 90.19 % / Multiplicity: 6.3 / Num. of structure factors: 17031 / Phase residual: 1.0E-6 ° |
| EM diffraction stats | Details: There was no phase error rejection criteria used for diffraction intensities. Fourier space coverage: 90.19 % / High resolution: 2.3 Å / Num. of intensities measured: 127703 / Num. of structure factors: 17031 / Phase error rejection criteria: 0 / Rmerge: 19.9 / Rsym: 13.8 |
| Reflection | Biso Wilson estimate: 31.17 Å2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal symmetry | Image processing-ID: 1 / ∠γ: 90 ° / C sampling length: 200 Å / A: 65.5 Å / B: 65.5 Å / C: 200 Å / Space group name H-M: P422
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CTF correction | Type: NONE | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.5 Å / Resolution method: DIFFRACTION PATTERN/LAYERLINES / Symmetry type: 2D CRYSTAL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 2B6O Pdb chain-ID: A / Accession code: 2B6O / Chain residue range: 6-255 / Details: Molecular replacement / Pdb chain residue range: 6-255 / Source name: PDB / Type: experimental model | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.5→2.5 Å / SU ML: 0.3818 / Cross valid method: FREE R-VALUE / σ(F): 1.33 / Phase error: 32.4947 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.15 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|

