 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8bmd: Structure of GroEL-ATP complex under continuous turnover conditions -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8bmd | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of GroEL-ATP complex under continuous turnover conditions | ||||||||||||
 Components Components | Chaperonin GroEL | ||||||||||||
 Keywords Keywords |  CHAPERONE / GroEL CHAPERONE / GroEL | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationGroEL-GroES complex / chaperonin ATPase / virion assembly / chaperone cofactor-dependent protein refolding / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat ...GroEL-GroES complex / chaperonin ATPase / virion assembly / chaperone cofactor-dependent protein refolding / isomerase activity / ATP-dependent protein folding chaperone / response to radiation / unfolded protein binding / protein folding / response to heat / protein refolding / magnesium ion binding / ATP hydrolysis activity / ATP binding / membrane / identical protein binding / cytosolSimilarity search - Function | ||||||||||||
| Biological species |  Escherichia coli (E. coli) Escherichia coli (E. coli) | ||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.8 Å | ||||||||||||
 Authors Authors | Dhurandhar, M. / Torino, S. / Efremov, R. | ||||||||||||
| Funding support | European Union,  Belgium, 3items Belgium, 3items
| ||||||||||||
 Citation Citation | Journal: Nat Methods / Year: 2023 Title: Time-resolved cryo-EM using a combination of droplet microfluidics with on-demand jetting. Authors: Stefania Torino / Mugdha Dhurandhar / Annelore Stroobants / Raf Claessens / Rouslan G Efremov / Abstract: Single-particle cryogenic electron microscopy (cryo-EM) allows reconstruction of high-resolution structures of proteins in different conformations. Protein function often involves transient ...Single-particle cryogenic electron microscopy (cryo-EM) allows reconstruction of high-resolution structures of proteins in different conformations. Protein function often involves transient functional conformations, which can be resolved using time-resolved cryo-EM (trEM). In trEM, reactions are arrested after a defined delay time by rapid vitrification of protein solution on the EM grid. Despite the increasing interest in trEM among the cryo-EM community, making trEM samples with a time resolution below 100 ms remains challenging. Here we report the design and the realization of a time-resolved cryo-plunger that combines a droplet-based microfluidic mixer with a laser-induced generator of microjets that allows rapid reaction initiation and plunge-freezing of cryo-EM grids. Using this approach, a time resolution of 5 ms was achieved and the protein density map was reconstructed to a resolution of 2.1 Å. trEM experiments on GroEL:GroES chaperonin complex resolved the kinetics of the complex formation and visualized putative short-lived conformations of GroEL-ATP complex. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8bmd.cif.gz | 1.3 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8bmd.ent.gz | 1.1 MB | Display | PDB format |
| PDBx/mmJSON format | 8bmd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bm/8bmdftp://data.pdbj.org/pub/pdb/validation_reports/bm/8bmd | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  16118MC  8bk7C  8bk8C  8bk9C  8bkaC  8bkbC  8bkgC  8bkzC  8bl2C  8bl7C  8blcC  8bldC  8bleC  8blfC  8blyC  8bm0C  8bm1C  8bmoC  8bmtC C: citing same article ( M: map data used to model this data |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 57391.711 Da / Num. of mol.: 14 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Escherichia coli (E. coli) / Gene: groEL, groL, mopA, b4143, JW4103 / Production host: Escherichia coli (E. coli) / References: UniProt: P0A6F5, chaperonin ATPase#2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: Mg#3: Chemical | ChemComp-ATP / Adenosine triphosphate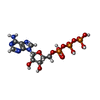  Mass: 507.181 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM Mass: 507.181 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM#4: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: K#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 1184 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1184 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: GroEL in complex with ATP under continuous turnover of ATP Type: COMPLEX / Entity ID: #1 / Source: RECOMBINANT | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.8 MDa / Experimental value: NO | ||||||||||||||||||||||||||||||
| Source (natural) | Organism: Escherichia coli (E. coli) / Cellular location: cytoplasm | ||||||||||||||||||||||||||||||
| Source (recombinant) | Organism: Escherichia coli (E. coli) | ||||||||||||||||||||||||||||||
| Buffer solution | pH: 7.5 | ||||||||||||||||||||||||||||||
| Buffer component |
| ||||||||||||||||||||||||||||||
| Specimen | Conc.: 3.7 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | ||||||||||||||||||||||||||||||
| Specimen support | Grid material: COPPER / Grid mesh size: 300 divisions/in. / Grid type: Quantifoil R2/1 | ||||||||||||||||||||||||||||||
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: JEOL CRYO ARM 300 |
|---|---|
| Electron gun | Electron source: FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELDBright-field microscopy / Nominal magnification: 60000 X / Nominal defocus max: 5000 nm / Nominal defocus min: 3500 nm / Cs: 2.55 mm / C2 aperture diameter: 100 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: JEOL CRYOSPECPORTER / Temperature (min): 100 K |
| Image recording | Electron dose: 63.6 e/Å2 / Film or detector model: GATAN K3 (6k x 4k) |
| EM imaging optics | Energyfilter name: In-column Omega Filter |
- Processing
Processing
| Software | Name: PHENIX / Version: 1.20.1_4487: / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software |
| ||||||||||||||||||||||||||||||||||||||||
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: D7 (2x7 fold dihedral) | ||||||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.8 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 20392 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||||||
| Atomic model building | Protocol: FLEXIBLE FIT / Space: REAL | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
