 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6ve5: X-ray structure of human REV7 in complex with Shieldin3 (residues... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ve5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray structure of human REV7 in complex with Shieldin3 (residues 41-74) | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  PROTEIN BINDING / DNA damage response / DNA double-strand break repair / DNA end resection / homologous recombination / non-homologous end joining / Shieldin3 / SHLD3 / RINN1 / REV7 / 53BP1 / RIF1 PROTEIN BINDING / DNA damage response / DNA double-strand break repair / DNA end resection / homologous recombination / non-homologous end joining / Shieldin3 / SHLD3 / RINN1 / REV7 / 53BP1 / RIF1 | ||||||
| Function / homology |  Function and homology information Function and homology informationsomatic diversification of immunoglobulins involved in immune response / DNA damage response, signal transduction resulting in transcription / telomere maintenance in response to DNA damage / negative regulation of transcription regulatory region DNA binding / zeta DNA polymerase complex / positive regulation of isotype switching / positive regulation of extracellular matrix assembly / negative regulation of epithelial to mesenchymal transition / negative regulation of transcription by competitive promoter binding / negative regulation of cell-cell adhesion mediated by cadherin ...somatic diversification of immunoglobulins involved in immune response / DNA damage response, signal transduction resulting in transcription / telomere maintenance in response to DNA damage / negative regulation of transcription regulatory region DNA binding / zeta DNA polymerase complex / positive regulation of isotype switching / positive regulation of extracellular matrix assembly / negative regulation of epithelial to mesenchymal transition / negative regulation of transcription by competitive promoter binding / negative regulation of cell-cell adhesion mediated by cadherin / JUN kinase binding / negative regulation of ubiquitin protein ligase activity / mitotic spindle assembly checkpoint signaling / positive regulation of double-strand break repair via nonhomologous end joining / negative regulation of double-strand break repair via homologous recombination / error-prone translesion synthesis / positive regulation of epithelial to mesenchymal transition / Translesion synthesis by REV1 / Translesion synthesis by POLK / Translesion synthesis by POLI / actin filament organization / regulation of cell growth / negative regulation of canonical Wnt signaling pathway / negative regulation of DNA-binding transcription factor activity / spindle / negative regulation of protein catabolic process / double-strand break repair / positive regulation of peptidyl-serine phosphorylation / site of double-strand break / chromosome / RNA polymerase II-specific DNA-binding transcription factor binding / cell division / DNA repair / chromatin / nucleolus / positive regulation of DNA-templated transcription / negative regulation of transcription by RNA polymerase II / nucleoplasm / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Cui, G. / Botuyan, M.V. / Mer, G. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: J Biol Chem / Year: 2021 Title: Cryo-EM reveals conformational flexibility in apo DNA polymerase ζ. Authors: Chloe Du Truong / Theodore A Craig / Gaofeng Cui / Maria Victoria Botuyan / Rachel A Serkasevich / Ka-Yi Chan / Georges Mer / Po-Lin Chiu / Rajiv Kumar / Abstract: The translesion synthesis (TLS) DNA polymerases Rev1 and Polζ function together in DNA lesion bypass during DNA replication, acting as nucleotide inserter and extender polymerases, respectively. ...The translesion synthesis (TLS) DNA polymerases Rev1 and Polζ function together in DNA lesion bypass during DNA replication, acting as nucleotide inserter and extender polymerases, respectively. While the structural characterization of the Saccharomyces cerevisiae Polζ in its DNA-bound state has illuminated how this enzyme synthesizes DNA, a mechanistic understanding of TLS also requires probing conformational changes associated with DNA- and Rev1 binding. Here, we used single-particle cryo-electron microscopy to determine the structure of the apo Polζ holoenzyme. We show that compared with its DNA-bound state, apo Polζ displays enhanced flexibility that correlates with concerted motions associated with expansion of the Polζ DNA-binding channel upon DNA binding. We also identified a lysine residue that obstructs the DNA-binding channel in apo Polζ, suggesting a gating mechanism. The Polζ subunit Rev7 is a hub protein that directly binds Rev1 and is a component of several other protein complexes such as the shieldin DNA double-strand break repair complex. We analyzed the molecular interactions of budding yeast Rev7 in the context of Polζ and those of human Rev7 in the context of shieldin using a crystal structure of Rev7 bound to a fragment of the shieldin-3 protein. Overall, our study provides new insights into Polζ mechanism of action and the manner in which Rev7 recognizes partner proteins. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ve5.cif.gz | 167.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ve5.ent.gz | 120.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6ve5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ve/6ve5ftp://data.pdbj.org/pub/pdb/validation_reports/ve/6ve5 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7lxdC  3abdS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24630.719 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: MAD2L2, MAD2B, REV7 / Plasmid: pETDuet-1 / Production host:  Escherichia coli (E. coli) / References: UniProt: Q9UI95 Escherichia coli (E. coli) / References: UniProt: Q9UI95 | ||||
|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 4067.683 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SHLD3, FLJ26957, RINN1 / Plasmid: pETDuet-1 / Production host: Escherichia coli BL21(DE3) (bacteria) / References: UniProt: Q6ZNX1 | ||||
| #3: Chemical | Sulfate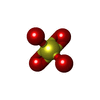  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 / Feature type: SUBJECT OF INVESTIGATION Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 48.7 % |
|---|---|
| Crystal grow | Temperature: 288 K / Method: vapor diffusion / pH: 6.5 Details: Protein complex was in 5 mM HEPES, pH 7.4, 100 mM NaCl, 5 mM DDT at 25 mg/mL;Reservoir solution was 0.1 M MES monohydrate, pH 6.5, 1.4 M MgSO4.6H20;Cryoprotection was done in 50% PEG 400 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 19-BM / Wavelength: 0.9794 Å |
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: Feb 15, 2019 / Details: Sagittal focusing 2nd crystal |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9794 Å / Relative weight: 1 |
| Reflection | Resolution: 2→29.25 Å / Num. obs: 19399 / % possible obs: 98.4 % / Redundancy: 22.6 % / Biso Wilson estimate: 24.07 Å2 / CC1/2: 0.999 / CC star: 1 / Rmerge(I) obs: 0.08271 / Rpim(I) all: 0.01825 / Rrim(I) all: 0.08478 / Net I/σ(I): 40.89 |
| Reflection shell | Resolution: 2→2.05 Å / Rmerge(I) obs: 1.193 / Mean I/σ(I) obs: 4.11 / Num. unique obs: 1337 / CC1/2: 0.847 / CC star: 0.958 / Rpim(I) all: 0.2621 / Rrim(I) all: 1.222 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3ABD Resolution: 2→29.25 Å / SU ML: 0.2082 / Cross valid method: FREE R-VALUE / σ(F): 1.35 / Phase error: 24.0879
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.57 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→29.25 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
