 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5zt1: Structure of the bacterial pathogens ATPase with substrate ATP gamma S -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5zt1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the bacterial pathogens ATPase with substrate ATP gamma S | |||||||||
 Components Components | Probable ATP synthase SpaL/MxiB | |||||||||
 Keywords Keywords |  HYDROLASE / ATPase / T3SS / hexamer / ATP gamma S HYDROLASE / ATPase / T3SS / hexamer / ATP gamma S | |||||||||
| Function / homology |  Function and homology information Function and homology informationprotein-exporting ATPase activity / protein-secreting ATPase / type III protein secretion system complex / protein secretion by the type III secretion system / biosynthetic process / ATP metabolic process / proton transmembrane transport / ATP hydrolysis activity / ATP binding / identical protein binding / cytoplasmSimilarity search - Function | |||||||||
| Biological species |  Shigella flexneri (bacteria) Shigella flexneri (bacteria) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.114 Å | |||||||||
 Authors Authors | Gao, X.P. / Mu, Z.X. / Cui, S. | |||||||||
| Funding support |  China, 2items China, 2items
| |||||||||
 Citation Citation | Journal: Front Microbiol / Year: 2018 Title: Structural Insight Into Conformational Changes Induced by ATP Binding in a Type III Secretion-Associated ATPase FromShigella flexneri Authors: Gao, X. / Mu, Z. / Yu, X. / Qin, B. / Wojdyla, J. / Wang, M. / Cui, S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5zt1.cif.gz | 259.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5zt1.ent.gz | 213.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5zt1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zt/5zt1ftp://data.pdbj.org/pub/pdb/validation_reports/zt/5zt1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5ybhC  5ybiC  2oblS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41181.637 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Shigella flexneri (bacteria) / Gene: spaL, mxiB, spa47, CP0149 / Plasmid: pET28A / Production host: Escherichia coli (E. coli) / Strain (production host): B834References: UniProt: P0A1C1, H+-transporting two-sector ATPase#2: Chemical | Sulfate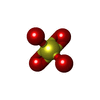  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 / Feature type: SUBJECT OF INVESTIGATION Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 29 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 29 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION#4: Chemical | ChemComp-AGS / | 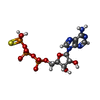  Mass: 523.247 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O12P3S / Feature type: SUBJECT OF INVESTIGATION / Comment: ATP-gamma-S, energy-carrying molecule analogue*YM Mass: 523.247 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O12P3S / Feature type: SUBJECT OF INVESTIGATION / Comment: ATP-gamma-S, energy-carrying molecule analogue*YM |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density meas: 0.01 Mg/m3 / Density % sol: 54 % Description: THE ENTRY CONTAINS FRIEDEL PAIRS IN F_PLUS/MINUS COLUMNS. |
|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion, hanging drop / pH: 8.6 / Details: 0.1M Bicine pH8.6;1.2M Ammonium sulphate |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL17U1 / Wavelength: 0.97876 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 24, 2013 |
| Radiation | Monochromator: Double Crystal Type Si (111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97876 Å / Relative weight: 1 |
| Reflection | Resolution: 3.1→50 Å / Num. obs: 16927 / % possible obs: 99.3 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 9.43 % / Rmerge(I) obs: 0.16 / Net I/σ(I): 17.78 |
| Reflection shell | Resolution: 3.11→3.3 Å / Redundancy: 8.86 % / Rmerge(I) obs: 0.75 / Mean I/σ(I) obs: 5.23 / % possible all: 96.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2obl Resolution: 3.114→29.868 Å / SU ML: 0.39 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 24.78 Details: Authors state that the low occupancy of the ligands result from only fewer protein molecules bound ligands.we balanced B-factors and occupancy of ligands when refinement. SF FILE CONTAINS ...Details: Authors state that the low occupancy of the ligands result from only fewer protein molecules bound ligands.we balanced B-factors and occupancy of ligands when refinement. SF FILE CONTAINS FRIEDEL PAIRS UNDER I/F_MINUS AND I/F_PLUS COLUMNS.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.114→29.868 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
