 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5lk4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the Red Fluorescent Protein mScarlet at pH 7.8 | |||||||||
 Components Components | mScarlet | |||||||||
 Keywords Keywords |  FLUORESCENT PROTEIN / beta barrel / red fluorescent protein FLUORESCENT PROTEIN / beta barrel / red fluorescent protein | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |  Discosoma sp. (sea anemone) Discosoma sp. (sea anemone) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 1.47 Å | |||||||||
 Authors Authors | Aumonier, S. / Gotthard, G. / Royant, A. | |||||||||
 Citation Citation | Journal: Nat. Methods / Year: 2017 Title: mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Authors: Bindels, D.S. / Haarbosch, L. / van Weeren, L. / Postma, M. / Wiese, K.E. / Mastop, M. / Aumonier, S. / Gotthard, G. / Royant, A. / Hink, M.A. / Gadella, T.W. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5lk4.cif.gz | 118.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5lk4.ent.gz | 90.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5lk4.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lk/5lk4ftp://data.pdbj.org/pub/pdb/validation_reports/lk/5lk4 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3kcsS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 24897.078 Da / Num. of mol.: 1 / Fragment: Red Fluorescent Protein mScarlet Source method: isolated from a genetically manipulated source Source: (gene. exp.) Discosoma sp. (sea anemone) / Production host:  Escherichia coli BL21(DE3) (bacteria) / References: UniProt: Q9U6Y8 Escherichia coli BL21(DE3) (bacteria) / References: UniProt: Q9U6Y8 |
|---|
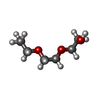
-Non-polymers , 5 types, 203 molecules 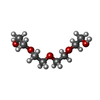
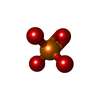


| #2: Chemical | ChemComp-PG4 / Polyethylene glycol Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM | ||||
|---|---|---|---|---|---|
| #3: Chemical | ChemComp-PO4 / Phosphate Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 | ||||
| #4: Chemical | ChemComp-PGE / Polyethylene glycol Mass: 150.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H14O4#5: Chemical | ChemComp-PEG / | Diethylene glycol Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 196 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.44 Å3/Da / Density % sol: 49.59 % Description: mScarlet crystal was soaked 45 min in 40% PEG 300, 100mM Sodium Phosphate Citrate pH 7.8 |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.2 / Details: 35-45% PEG 300, 100mM Sodium Phosphate Citrate / PH range: 4.0 - 5.0 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.978 Å / Beamline: ID29 / Wavelength: 0.978 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: May 18, 2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Si [111] / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.978 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.47→81 Å / Num. obs: 40759 / % possible obs: 98.6 % / Observed criterion σ(I): -3 / Redundancy: 3.3 % / Biso Wilson estimate: 20.552 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.065 / Net I/σ(I): 10.99 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: pdbid 3KCS Resolution: 1.47→80.98 Å / Cor.coef. Fo:Fc: 0.977 / Cor.coef. Fo:Fc free: 0.967 / SU B: 2.397 / SU ML: 0.039 / Cross valid method: FREE R-VALUE / σ(F): 0 / ESU R: 0.068 / ESU R Free: 0.059 / Details: REFMAC 5.8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 76.5 Å2 / Biso mean: 16.993 Å2 / Biso min: 8.79 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.47→80.98 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.47→1.508 Å / Total num. of bins used: 20
|
