 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5dcu: Iridoid synthase from Catharanthus roseus - ternary complex with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5dcu | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Iridoid synthase from Catharanthus roseus - ternary complex with NADP+ and triethylene glycol carboxylic acid | ||||||||||||
 Components Components | Iridoid synthase | ||||||||||||
 Keywords Keywords |  OXIDOREDUCTASE / Iridoid synthase / short chain dehydrogenase / NADPH-dependent / Catharanthus roseus OXIDOREDUCTASE / Iridoid synthase / short chain dehydrogenase / NADPH-dependent / Catharanthus roseus | ||||||||||||
| Function / homology |  Function and homology information Function and homology information(S)-8-oxocitronellyl enol synthase / monoterpenoid biosynthetic process / oxidoreductase activity, acting on the CH-CH group of donors, NAD or NADP as acceptor / protein homodimerization activity / identical protein binding / cytosolSimilarity search - Function | ||||||||||||
| Biological species |  Catharanthus roseus (Madagascar periwinkle) Catharanthus roseus (Madagascar periwinkle) | ||||||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.4 Å | ||||||||||||
 Authors Authors | Caputi, L. / Kries, H. / Stevenson, C.E.M. / Kamileen, M.O. / Sherden, N.H. / Geu-Flores, F. / Lawson, D.M. / O'Connor, S.E. | ||||||||||||
| Funding support |  United Kingdom, United Kingdom,  Switzerland, 3items Switzerland, 3items
| ||||||||||||
 Citation Citation | Journal: Nat.Chem.Biol. / Year: 2016 Title: Structural determinants of reductive terpene cyclization in iridoid biosynthesis. Authors: Kries, H. / Caputi, L. / Stevenson, C.E. / Kamileen, M.O. / Sherden, N.H. / Geu-Flores, F. / Lawson, D.M. / O'Connor, S.E. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5dcu.cif.gz | 331.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5dcu.ent.gz | 270.1 KB | Display | PDB format |
| PDBx/mmJSON format | 5dcu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dc/5dcuftp://data.pdbj.org/pub/pdb/validation_reports/dc/5dcu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5dcwC  5dcyC  5df1C  2v6gS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Components on special symmetry positions |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 0 / Ens-ID: 1 / Beg auth comp-ID: GLY / Beg label comp-ID: GLY / End auth comp-ID: PHE / End label comp-ID: PHE / Refine code: 0 / Auth seq-ID: 23 - 391 / Label seq-ID: 3 - 371
|
-Components
| #1: Protein | Mass: 41771.969 Da / Num. of mol.: 2 / Fragment: UNP residues 23-388 Source method: isolated from a genetically manipulated source Details: The crystallised protein contained residues 23-388 of the wild-type amino acid sequence. The sequence differed from database entry K7WDL7 by an Asp to Asn change at position 87. The N- ...Details: The crystallised protein contained residues 23-388 of the wild-type amino acid sequence. The sequence differed from database entry K7WDL7 by an Asp to Asn change at position 87. The N-terminus retained two residues from the nickel affinity cleavage site. The C-terminus had an additional three vector-derived residues. Source: (gene. exp.) Catharanthus roseus (Madagascar periwinkle)Plasmid: pOPIN-F / Production host:  Escherichia coli BL21(DE3) (bacteria) / Variant (production host): Rosetta 2 / References: UniProt: K7WDL7, EC: 1.3.1.99 Escherichia coli BL21(DE3) (bacteria) / Variant (production host): Rosetta 2 / References: UniProt: K7WDL7, EC: 1.3.1.99#2: Chemical | Nicotinamide adenine dinucleotide phosphate  Mass: 743.405 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H28N7O17P3#3: Chemical | ChemComp-EDO / Ethylene glycol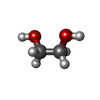  Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#4: Chemical |   Mass: 164.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H12O5 Mass: 164.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H12O5#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 852 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 852 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.16 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04 / Wavelength: 0.9 Å | |||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jul 9, 2014 | |||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.9 Å / Relative weight: 1 | |||||||||||||||||||||||||||
| Reflection | Resolution: 1.4→86.32 Å / Num. obs: 149766 / % possible obs: 100 % / Redundancy: 8.3 % / Biso Wilson estimate: 11.4 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.069 / Rpim(I) all: 0.025 / Net I/σ(I): 16.1 / Num. measured all: 1249645 | |||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1 / Rejects: 0
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2V6G Resolution: 1.4→86.32 Å / Cor.coef. Fo:Fc: 0.976 / Cor.coef. Fo:Fc free: 0.966 / WRfactor Rfree: 0.172 / WRfactor Rwork: 0.148 / FOM work R set: 0.8783 / SU B: 2.005 / SU ML: 0.04 / SU R Cruickshank DPI: 0.055 / SU Rfree: 0.0571 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.055 / ESU R Free: 0.057 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.7 Å / Shrinkage radii: 0.7 Å / VDW probe radii: 1.1 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 60.16 Å2 / Biso mean: 19.9 Å2 / Biso min: 9.94 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.4→86.32 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Ens-ID: 1 / Number: 46448 / Refine-ID: X-RAY DIFFRACTION / Type: interatomic distance / Rms dev position: 0.07 Å / Weight position: 0.05
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.4→1.436 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
