- PDB-4rh7: Crystal structure of human cytoplasmic dynein 2 motor domain in c... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4rh7
Title
Crystal structure of human cytoplasmic dynein 2 motor domain in complex with ADP.Vi
Components
Green fluorescent protein/Cytoplasmic dynein 2 heavy chain 1
Keywords
MOTOR PROTEIN / AAA+ protein / dynein motor domain
Function / homology
Function and homology information
intraciliary retrograde transport / cilium movement involved in cell motility / 9+2 motile cilium / spinal cord motor neuron differentiation / ciliary tip / Intraflagellar transport / protein localization to cilium / minus-end-directed microtubule motor activity / cytoplasmic dynein complex / non-motile cilium assembly ...intraciliary retrograde transport / cilium movement involved in cell motility / 9+2 motile cilium / spinal cord motor neuron differentiation / ciliary tip / Intraflagellar transport / protein localization to cilium / minus-end-directed microtubule motor activity / cytoplasmic dynein complex / non-motile cilium assembly / coronary vasculature development / dynein light intermediate chain binding / positive regulation of smoothened signaling pathway / dorsal/ventral pattern formation / determination of left/right symmetry / embryonic limb morphogenesis / dynein intermediate chain binding / Golgi organization / axoneme / cytoskeletal motor activity / Hedgehog 'off' state / forebrain development / kidney development / cilium / protein processing / apical part of cell / microtubule / Golgi apparatus / ATP hydrolysis activity / extracellular exosome / ATP binding / plasma membrane Similarity search - Function
: / Cytoplasmic dynein 2 heavy chain 1, AAA+ ATPase domain / Dynein heavy chain, C-terminal domain / Dynein heavy chain, C-terminal domain, barrel region / Dynein heavy chain C-terminal domain / P-loop containing dynein motor region / Dynein heavy chain, tail / Dynein heavy chain, N-terminal region 1 / Dynein heavy chain / Dynein heavy chain region D6 P-loop domain ...: / Cytoplasmic dynein 2 heavy chain 1, AAA+ ATPase domain / Dynein heavy chain, C-terminal domain / Dynein heavy chain, C-terminal domain, barrel region / Dynein heavy chain C-terminal domain / P-loop containing dynein motor region / Dynein heavy chain, tail / Dynein heavy chain, N-terminal region 1 / Dynein heavy chain / Dynein heavy chain region D6 P-loop domain / Dynein heavy chain, linker / Dynein heavy chain, AAA module D4 / Dynein heavy chain, coiled coil stalk / Dynein heavy chain, hydrolytic ATP-binding dynein motor region / Dynein heavy chain, ATP-binding dynein motor region / Dynein heavy chain AAA lid domain / Dynein heavy chain AAA lid domain superfamily / Dynein heavy chain, domain 2, N-terminal / Dynein heavy chain, linker, subdomain 3 / Dynein heavy chain, AAA1 domain, small subdomain / Dynein heavy chain region D6 P-loop domain / Dynein heavy chain, N-terminal region 2 / Hydrolytic ATP binding site of dynein motor region / Microtubule-binding stalk of dynein motor / P-loop containing dynein motor region D4 / ATP-binding dynein motor region / Dynein heavy chain AAA lid domain / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 5.83 Å3/Da / Density % sol: 78.89 %
Crystal grow
Temperature: 292 K / Method: vapor diffusion, hanging drop Details: mixing equal volumes of protein (8mg/ml) and reservoir solution (4-6 % PEG 6000 and 0.1 M Tris pH 8.0), VAPOR DIFFUSION, HANGING DROP, temperature 292K
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Relative weight: 1
Reflection
Resolution: 3.4→56.5 Å / Num. obs: 78411 / % possible obs: 62.2 % / Redundancy: 4.1 % / Rsym value: 0.101 / Net I/σ(I): 7.6
-
Processing
Software
Name
Version
Classification
SHARP
phasing
REFMAC
5.8.0073
refinement
MOSFLM
datareduction
SCALA
datascaling
Refinement
Method to determine structure: MIRAS / Resolution: 3.41→56.6 Å / Cor.coef. Fo:Fc: 0.924 / Cor.coef. Fo:Fc free: 0.886 / SU B: 35.363 / SU ML: 0.532 / Cross valid method: THROUGHOUT / ESU R Free: 0.643 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.28546
3915
5 %
RANDOM
Rwork
0.23746
-
-
-
obs
0.23989
74060
62.16 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords MOTOR PROTEIN /
MOTOR PROTEIN /  Function and homology information
Function and homology information
 Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
 PDBj
PDBj




 Assembly
Assembly


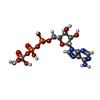
 Mass: 544.156 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N5O14P2V / Comment: energy-carrying molecule analogue*YM
Mass: 544.156 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N5O14P2V / Comment: energy-carrying molecule analogue*YM
 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg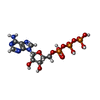
 Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM
Mass: 507.181 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H16N5O13P3 / Comment: ATP, energy-carrying molecule*YM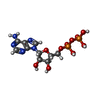
 Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM
Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Sample preparation
Sample preparation / Beamline: I02
/ Beamline: I02 Processing
Processing