 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4ffb: A TOG:alpha/beta-tubulin Complex Structure Reveals Conformation-B... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4ffb | ||||||
|---|---|---|---|---|---|---|---|
| Title | A TOG:alpha/beta-tubulin Complex Structure Reveals Conformation-Based Mechanisms For a Microtubule Polymerase | ||||||
 Components Components |
| ||||||
 Keywords Keywords |  HYDROLASE / tubulin fold / HEAT repeats / cytoskeleton / microtubule / tubulin / TOG domain HYDROLASE / tubulin fold / HEAT repeats / cytoskeleton / microtubule / tubulin / TOG domain | ||||||
| Function / homology |  Function and homology information Function and homology informationrepair of mitotic kinetochore microtubule attachment defect / microtubule plus end polymerase / Cilium Assembly / nuclear migration by microtubule mediated pushing forces / mitotic sister chromatid biorientation / establishment or maintenance of microtubule cytoskeleton polarity / nuclear division / Sealing of the nuclear envelope (NE) by ESCRT-III / mitotic spindle elongation / nuclear migration along microtubule ...repair of mitotic kinetochore microtubule attachment defect / microtubule plus end polymerase / Cilium Assembly / nuclear migration by microtubule mediated pushing forces / mitotic sister chromatid biorientation / establishment or maintenance of microtubule cytoskeleton polarity / nuclear division / Sealing of the nuclear envelope (NE) by ESCRT-III / mitotic spindle elongation / nuclear migration along microtubule / homologous chromosome segregation / Platelet degranulation / positive regulation of intracellular protein transport / microtubule plus-end binding / spindle pole body / tubulin complex / microtubule polymerization / mitotic sister chromatid segregation / mitotic spindle assembly / microtubule-based process / cytoplasmic microtubule organization / cytoskeleton organization / Neutrophil degranulation / nuclear periphery / mitotic spindle organization / Hydrolases; Acting on acid anhydrides; Acting on GTP to facilitate cellular and subcellular movement / spindle microtubule / structural constituent of cytoskeleton / spindle / kinetochore / microtubule cytoskeleton organization / spindle pole / mitotic cell cycle / cell cortex / microtubule binding / microtubule / hydrolase activity / response to antibiotic / GTPase activity / GTP binding / metal ion binding / nucleus / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.882 Å | ||||||
 Authors Authors | Ayaz, P. / Ye, X. / Huddleston, P. / Brautigam, C.A. / Rice, L.M. | ||||||
 Citation Citation | Journal: Science / Year: 2012 Title: A TOG: alpha beta-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Authors: Ayaz, P. / Ye, X. / Huddleston, P. / Brautigam, C.A. / Rice, L.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4ffb.cif.gz | 440.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4ffb.ent.gz | 361 KB | Display | PDB format |
| PDBx/mmJSON format | 4ffb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ff/4ffbftp://data.pdbj.org/pub/pdb/validation_reports/ff/4ffb | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 49853.867 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Strain: ATCC 204508 / S288c / Gene: TUB1, YML085C / Plasmid: p426Gal1 / Production host: Saccharomyces cerevisiae (brewer's yeast) / Strain (production host): JEL1 / References: UniProt: P09733 | ||
|---|---|---|---|
| #2: Protein | Mass: 51910.477 Da / Num. of mol.: 1 / Mutation: T175R,V179R Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Strain: ATCC 204508 / S288c / Gene: TUB2, YFL037W / Plasmid: p424Gal1 / Production host: Saccharomyces cerevisiae (brewer's yeast) / Strain (production host): JEL1 / References: UniProt: P02557, EC: 3.6.5.6 | ||
| #3: Protein | Mass: 31666.191 Da / Num. of mol.: 1 / Fragment: TOG1 domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Saccharomyces cerevisiae (brewer's yeast)Strain: ATCC 204508 / S288c / Gene: L2108, STU2, YLR045C / Plasmid: pET28 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P46675 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P46675 | ||
| #4: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#5: Chemical | Guanosine triphosphate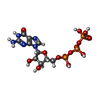  Mass: 523.180 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM Mass: 523.180 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.95 Å3/Da / Density % sol: 58.29 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 5.9 Details: 20% (v/v) PEG 400, 0.1 M MES pH 5.9, VAPOR DIFFUSION, SITTING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.9794 Å / Beamline: 19-ID / Wavelength: 0.9794 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Nov 17, 2010 |
| Radiation | Monochromator: SAGITALLY FOCUSED Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9794 Å / Relative weight: 1 |
| Reflection | Resolution: 2.88→50 Å / Num. all: 39347 / Num. obs: 39347 / % possible obs: 86 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 |
| Reflection shell | Resolution: 2.88→2.95 Å / Redundancy: 3.2 % / Rmerge(I) obs: 0.967 / Mean I/σ(I) obs: 0.9 / % possible all: 22.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1SA0 CHAIN A, 1SA0 CHAIN B, 2QK1 Resolution: 2.882→44.945 Å / SU ML: 0.38 / σ(F): 1.35 / Phase error: 29.61 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.882→44.945 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
