 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3l1g | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human AlphaB crystallin | ||||||
 Components Components | Alpha-crystallin B chain CRYAB CRYAB | ||||||
 Keywords Keywords | CHAPERONE / lens transparency / polydispersity / protein aggregation / crystallin / Eye lens protein | ||||||
| Function / homology |  Function and homology information Function and homology informationmicrotubule polymerization or depolymerization / negative regulation of intracellular transport / apoptotic process involved in morphogenesis / cardiac myofibril / regulation of programmed cell death / tubulin complex assembly / structural constituent of eye lens / negative regulation of amyloid fibril formation / M band / lens development in camera-type eye ...microtubule polymerization or depolymerization / negative regulation of intracellular transport / apoptotic process involved in morphogenesis / cardiac myofibril / regulation of programmed cell death / tubulin complex assembly / structural constituent of eye lens / negative regulation of amyloid fibril formation / M band / lens development in camera-type eye / muscle organ development / actin filament bundle / HSF1-dependent transactivation / negative regulation of reactive oxygen species metabolic process / negative regulation of protein-containing complex assembly / stress-activated MAPK cascade / muscle contraction / synaptic membrane / response to hydrogen peroxide / cellular response to gamma radiation / negative regulation of cell growth / Z disc / unfolded protein binding / protein folding / response to estradiol / amyloid-beta binding / response to heat / perikaryon / protein refolding / microtubule binding / dendritic spine / lysosome / response to hypoxia / protein stabilization / axon / negative regulation of gene expression / negative regulation of DNA-templated transcription / protein-containing complex binding / negative regulation of apoptotic process / structural molecule activity / cell surface / protein homodimerization activity / protein-containing complex / mitochondrion / extracellular exosome / nucleoplasm / identical protein binding / metal ion binding / nucleus / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.32 Å | ||||||
 Authors Authors | Laganowsky, A. / Sawaya, M.R. / Cascio, D. / Eisenberg, D. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2010 Title: Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Authors: Laganowsky, A. / Benesch, J.L. / Landau, M. / Ding, L. / Sawaya, M.R. / Cascio, D. / Huang, Q. / Robinson, C.V. / Horwitz, J. / Eisenberg, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3l1g.cif.gz | 52.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3l1g.ent.gz | 38.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3l1g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l1/3l1gftp://data.pdbj.org/pub/pdb/validation_reports/l1/3l1g | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | CRYAB / Alpha(B)-crystallin / Rosenthal fiber component / Heat shock protein beta-5 / HspB5 / Renal ...Alpha(B)-crystallin / Rosenthal fiber component / Heat shock protein beta-5 / HspB5 / Renal carcinoma antigen NY-REN-27 Mass: 10923.354 Da / Num. of mol.: 1 / Fragment: residues 68-162 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CRYA2, CRYAB / Plasmid: pET28 / Production host:  Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P02511 Escherichia coli (E. coli) / Strain (production host): BL21(DE3) / References: UniProt: P02511 |
|---|---|
| #2: Chemical | Sulfate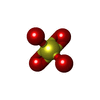  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.6 Å3/Da / Density % sol: 65.84 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 4.6 Details: 0.08M SODIUM ACETATE pH 4.6, 20% GLYCEROL, 1.8M AMMONIUM SULFATE, vapor diffusion, sitting drop, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 24-ID-C / Wavelength: 1.49 Å / Beamline: 24-ID-C / Wavelength: 1.49 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Aug 15, 2009 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.49 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 3.15→18.88 Å / Num. obs: 2777 / % possible obs: 98.3 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 93.901 Å2 / Rmerge(I) obs: 0.055 / Net I/σ(I): 19.05 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 3.32→18.878 Å / Occupancy max: 1 / Occupancy min: 0.5 / FOM work R set: 0.669 / SU ML: -0 / σ(F): 1.99 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 148.701 Å2 / ksol: 0.301 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 345.5 Å2 / Biso mean: 165.659 Å2 / Biso min: 20 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.32→18.878 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 2
| ||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 13.9412 Å / Origin y: -6.1514 Å / Origin z: 3.5444 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
