 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pw5 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | putative nagD protein | |||||||||
 Components Components | nagD protein, putative | |||||||||
 Keywords Keywords |  STRUCTURAL GENOMICS / UNKNOWN FUNCTION / nagD protein / T. maritima / PSI / Protein Structure Initiative / Midwest Center for Structural Genomics / MCSG STRUCTURAL GENOMICS / UNKNOWN FUNCTION / nagD protein / T. maritima / PSI / Protein Structure Initiative / Midwest Center for Structural Genomics / MCSG | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | |||||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 2.8 Å | |||||||||
 Authors Authors | Cuff, M.E. / Skarina, T. / Savchenko, A. / Edwards, A. / Joachimiak, A. / Midwest Center for Structural Genomics (MCSG) | |||||||||
 Citation Citation | Journal: To be Published Title: putative nagD protein Authors: Cuff, M.E. / Skarina, T. / Savchenko, A. / Edwards, A. / Joachimiak, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pw5.cif.gz | 61.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pw5.ent.gz | 48.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1pw5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pw/1pw5ftp://data.pdbj.org/pub/pdb/validation_reports/pw/1pw5 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data | |
|---|---|
| Other databases |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 28559.908 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (bacteria) / Gene: TM1742 / Production host: Escherichia coli (E. coli) / References: UniProt: Q9X264 | ||
|---|---|---|---|
| #2: Sugar | ChemComp-NDG / N-Acetylglucosamine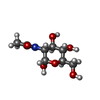  Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 | ||
| #3: Chemical | Sulfate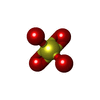  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: amonium sulfate, sodium cacodylate, ethylene glycol, pH 6.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
|---|
-Data collection
| Diffraction | Mean temperature: 150 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97929,0.97918,0.96396 / Beamline: 19-ID / Wavelength: 0.97929,0.97918,0.96396 | ||||||||||||
| Detector | Type: SBC-2 / Detector: CCD / Date: Jul 26, 2002 | ||||||||||||
| Radiation | Monochromator: double crystal Si(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.8→28.93 Å / Num. all: 18344 / Num. obs: 18344 / Observed criterion σ(F): 0 / Biso Wilson estimate: 70.6 Å2 / Limit h max: 34 / Limit h min: 0 / Limit k max: 24 / Limit k min: 0 / Limit l max: 56 / Limit l min: 0 / Observed criterion F max: 127621.03 / Observed criterion F min: 0.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.8→28.93 Å / Rfactor Rfree error: 0.01 / Occupancy max: 1 / Occupancy min: 1 / Cross valid method: THROUGHOUT Details: The authors state there is extra electron density near/in the main chain between residues A239 and A242. It could be an extra amino acid (cloning artifact), or some sort of ligand or ligands. ...Details: The authors state there is extra electron density near/in the main chain between residues A239 and A242. It could be an extra amino acid (cloning artifact), or some sort of ligand or ligands. The authors are confident of the chain trace.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: CNS bulk solvent model used / Bsol: 54.6016 Å2 / ksol: 0.297685 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 150.17 Å2 / Biso mean: 75.24 Å2 / Biso min: 29.03 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→28.93 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
