 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1oko: Crystal structure of Pseudomonas Aeruginosa Lectin 1 complexed wi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1oko | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Pseudomonas Aeruginosa Lectin 1 complexed with galactose at 1.6 A resolution | ||||||
 Components Components | PA-I GALACTOPHILIC LECTIN | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN /  GALACTOSE BINDING GALACTOSE BINDING | ||||||
| Function / homology |  Function and homology information Function and homology informationheterophilic cell-cell adhesion via plasma membrane cell adhesion molecules / carbohydrate binding / periplasmic space / cell surface / cytoplasmSimilarity search - Function | ||||||
| Biological species |   PSEUDOMONAS AERUGINOSA (bacteria) PSEUDOMONAS AERUGINOSA (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Cioci, G. / Mitchell, E. / Gautier, C. / Wimmerova, M. / Perez, S. / Gilboa-Garber, N. / Imberty, A. | ||||||
 Citation Citation | Journal: FEBS Lett. / Year: 2003 Title: Structural Basis of Calcium and Galactose Recognition by the Lectin Pa-Il of Pseudomonas Aeruginosa Authors: Cioci, G. / Mitchell, E. / Gautier, C. / Wimmerova, M. / Sudakevitz, D. / Perez, S. / Gilboa-Garber, N. / Imberty, A. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1oko.cif.gz | 114.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1oko.ent.gz | 89.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1oko.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ok/1okoftp://data.pdbj.org/pub/pdb/validation_reports/ok/1oko | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1uojC  1okpS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 4 molecules ABCD
| #1: Protein | Mass: 12770.137 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Source: (natural) PSEUDOMONAS AERUGINOSA (bacteria) / References: UniProt: Q05097 |
|---|
-Sugars , 2 types, 7 molecules 
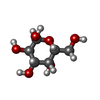
| #3: Sugar | ChemComp-GAL / Galactose Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 4 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H12O6 #6: Sugar | Galactose Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 3 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 3Source method: isolated from a genetically manipulated source Formula: C6H12O6 |
|---|
-Non-polymers , 4 types, 490 molecules 
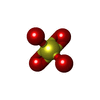


| #2: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#4: Chemical | Sulfate Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-MPD / ( | 2-Methyl-2,4-pentanediol Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 482 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2 Å3/Da / Density % sol: 37 % / Description: RFREE MERGED FROM EBI-13164 | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 5 Details: HANGING DROP: PROTEIN SOLUTION: PA1L 5 MG/ML, D-GAL 0.025 MG/ML, CACL2 & MGCL2 2MM RESERVOIR SOLUTION: (NH4)2SO4 1.5 M, PH 4.7, 5% MPD, 2% GLYCEROL 2 UL + 2 UL | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 4.7 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 / Beamline: ID14-1 / Wavelength: 0.934 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Jun 15, 2003 / Details: MULTILAYER |
| Radiation | Monochromator: DIAMOND / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→18.6 Å / Num. obs: 53762 / % possible obs: 94.9 % / Redundancy: 4.49 % / Biso Wilson estimate: 12.535 Å2 / Rmerge(I) obs: 0.076 / Net I/σ(I): 6.6701 |
| Reflection shell | Resolution: 1.6→1.66 Å / Redundancy: 4.23 % / Rmerge(I) obs: 0.41 / Mean I/σ(I) obs: 1.42 / % possible all: 79.1 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Redundancy: 4.5 % / Num. measured all: 241596 / Rmerge(I) obs: 0.076 |
| Reflection shell | *PLUS % possible obs: 79.1 % / Redundancy: 4.2 % / Rmerge(I) obs: 0.41 / Mean I/σ(I) obs: 1.4 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: MONOMER B FROM PREVIOULSY RESOLVED 1OKP Resolution: 1.6→18.602 Å / SU B: 1.779 / SU ML: 0.062 / Cross valid method: THROUGHOUT / ESU R: 0.091 / ESU R Free: 0.09
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 11.301 Å2
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→18.602 Å
| ||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 18.6 Å / Rfactor Rfree: 0.187 / Rfactor Rwork: 0.154 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.256 / Num. reflection Rfree: 144 / Rfactor Rwork: 0.197 / Num. reflection Rwork: 3066 |
