 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1o6t: Internalin (INLA, Listeria monocytogenes) - functional domain, un... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1o6t | ||||||
|---|---|---|---|---|---|---|---|
| Title | Internalin (INLA, Listeria monocytogenes) - functional domain, uncomplexed | ||||||
 Components Components | INTERNALIN A | ||||||
 Keywords Keywords | CELL INVASION /  BACTERIAL INFECTION / LEUCINE RICH REPEAT / CELL ADHESION / CELL-WALL SURFACE PROTEIN BACTERIAL INFECTION / LEUCINE RICH REPEAT / CELL ADHESION / CELL-WALL SURFACE PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationInlA-mediated entry of Listeria monocytogenes into host cells / peptidoglycan-based cell wall / extracellular region Similarity search - Function | ||||||
| Biological species |  LISTERIA MONOCYTOGENES (bacteria) LISTERIA MONOCYTOGENES (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Schubert, W.-D. / Urbanke, C. / Ziehm, T. / Beier, V. / Machner, M.P. / Domann, E. / Wehland, J. / Chakraborty, T. / Heinz, D.W. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2002 Title: Structure of Internalin, a Major Invasion Protein of Listeria Monocytogenes, in Complex with its Human Receptor E-Cadherin Authors: Schubert, W.-D. / Urbanke, C. / Ziehm, T. / Beier, V. / Machner, M.P. / Domann, E. / Wehland, J. / Chakraborty, T. / Heinz, D.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1o6t.cif.gz | 138.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1o6t.ent.gz | 108.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1o6t.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/o6/1o6tftp://data.pdbj.org/pub/pdb/validation_reports/o6/1o6t | HTTPS FTP |
|---|
-Related structure data








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 50239.133 Da / Num. of mol.: 1 / Fragment: FUNCTIONAL DOMAIN, RESIDUES 36-496 Source method: isolated from a genetically manipulated source Source: (gene. exp.) LISTERIA MONOCYTOGENES (bacteria) / Strain: EGD-E / Variant: SEROVAR 1/2A / Plasmid: PGEX-6P-1 / Production host: ESCHERICHIA COLI (E. coli) / Strain (production host): BL21 / References: UniProt: P25146, UniProt: P0DJM0*PLUS |
|---|
-Non-polymers , 6 types, 715 molecules 


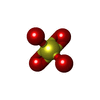


| #2: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||||||
|---|---|---|---|---|---|---|---|
| #3: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca | ||||||
| #4: Chemical | Chloride Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#5: Chemical | ChemComp-SO4 / Sulfate Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#6: Chemical | ChemComp-MES / MES (buffer) Mass: 195.237 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#7: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 702 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Sequence details | FIRST FIVE RESIDUES (GPLGS) INTRODUCED |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.42 Å3/Da / Density % sol: 48.8 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6 Details: METHOD: VAPOUR DIFFUSION / HANGING DROP, PROTEIN: 10 MG/ML IN 10 MM HEPES PH 7.0, RESEVOIR: 10% PEG 4000, 100 MM MES/TRIS PH 6.0, 100 MM AMMONIUM SULFATE, 10 MM CACL2. CRYSTALS GREW APPROX 2 ...Details: METHOD: VAPOUR DIFFUSION / HANGING DROP, PROTEIN: 10 MG/ML IN 10 MM HEPES PH 7.0, RESEVOIR: 10% PEG 4000, 100 MM MES/TRIS PH 6.0, 100 MM AMMONIUM SULFATE, 10 MM CACL2. CRYSTALS GREW APPROX 2 YEARS AFTER SETUP. TRUE CRYSTALLIZATION CONDITIONS PROBABLY DEVIATE FROM THOSE INDICATED | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1.05 / Beamline: BW6 / Wavelength: 1.05 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Aug 15, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.05 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→25.1 Å / Num. obs: 50336 / % possible obs: 92 % / Redundancy: 6.2 % / Rmerge(I) obs: 0.062 / Net I/σ(I): 14.4 |
| Reflection shell | Resolution: 1.8→1.86 Å / Redundancy: 4.5 % / Rmerge(I) obs: 0.385 / Mean I/σ(I) obs: 3.4 / % possible all: 84.3 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Lowest resolution: 20 Å / Num. obs: 62613 / % possible obs: 99.5 % / Redundancy: 3.6 % / Rmerge(I) obs: 0.048 |
| Reflection shell | *PLUS Highest resolution: 1.6 Å / Lowest resolution: 1.63 Å / % possible obs: 99.2 % / Redundancy: 3.5 % / Rmerge(I) obs: 0.317 / Mean I/σ(I) obs: 3.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.6→91.29 Å / Cor.coef. Fo:Fc: 0.977 / Cor.coef. Fo:Fc free: 0.969 / SU B: 1.384 / SU ML: 0.049 / Cross valid method: THROUGHOUT / ESU R: 0.077 / ESU R Free: 0.077 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.26 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→91.29 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|