 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gg6: CRYSTAL STRUCTURE OF GAMMA CHYMOTRYPSIN WITH N-ACETYL-PHENYLALANI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gg6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF GAMMA CHYMOTRYPSIN WITH N-ACETYL-PHENYLALANINE TRIFLUOROMETHYL KETONE BOUND AT THE ACTIVE SITE | ||||||
 Components Components | (GAMMA CHYMOTRYPSIN) x 3 | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR /  CHYMOTRYPSIN / HYDROLASE-HYDROLASE INHIBITOR COMPLEX CHYMOTRYPSIN / HYDROLASE-HYDROLASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology informationchymotrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / serine-type endopeptidase activity / proteolysis / extracellular region Function and homology informationchymotrypsin / serpin family protein binding / serine protease inhibitor complex / digestion / serine-type endopeptidase activity / proteolysis / extracellular regionSimilarity search - Function | ||||||
| Biological species |  Bos taurus (cattle) Bos taurus (cattle) | ||||||
| Method | X-RAY DIFFRACTION / Resolution: 1.4 Å | ||||||
 Authors Authors | Neidhart, D. / Wei, Y. / Cassidy, C. / Lin, J. / Cleland, W.W. / Frey, P.A. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2001 Title: Correlation of low-barrier hydrogen bonding and oxyanion binding in transition state analogue complexes of chymotrypsin. Authors: Neidhart, D. / Wei, Y. / Cassidy, C. / Lin, J. / Cleland, W.W. / Frey, P.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gg6.cif.gz | 68.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gg6.ent.gz | 49.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1gg6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gg/1gg6ftp://data.pdbj.org/pub/pdb/validation_reports/gg/1gg6 | HTTPS FTP |
|---|
-Related structure data














-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
-Protein/peptide , 1 types, 1 molecules A
| #1: Protein/peptide | Mass: 996.223 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Bos taurus (cattle) / Organ: PANCREAS / References: UniProt: P00766, chymotrypsin |
|---|
-Protein , 2 types, 2 molecules BC
| #2: Protein | Mass: 13934.556 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Bos taurus (cattle) / Organ: PANCREAS / References: UniProt: P00766, chymotrypsin |
|---|---|
| #3: Protein | Mass: 10074.495 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Bos taurus (cattle) / Organ: PANCREAS / References: UniProt: P00766, chymotrypsin |
-Non-polymers , 5 types, 342 molecules 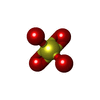

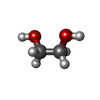


| #4: Chemical | Sulfate Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-APL / |  Mass: 277.240 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H14F3NO3 Mass: 277.240 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H14F3NO3#6: Chemical | ChemComp-EDO / Ethylene glycol Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#7: Chemical | ChemComp-APF / |  Mass: 259.224 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H12F3NO2 Mass: 259.224 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H12F3NO2#8: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 333 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.28 Å3/Da / Density % sol: 46.15 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion / pH: 7 Details: ammonium sulfate, potassium phosphate, pH 7.0, vapor diffusion, temperature 298.0K | |||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 113 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: SIEMENS / Detector: AREA DETECTOR / Date: Nov 29, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→30 Å / Num. all: 45082 / Num. obs: 42041 / % possible obs: 91.4 % / Observed criterion σ(I): 2 / Redundancy: 3.6 % / Rmerge(I) obs: 0.027 |
| Reflection shell | Resolution: 1.4→1.45 Å / Rmerge(I) obs: 0.118 / % possible all: 75.8 |
| Reflection | *PLUS Num. obs: 45082 |
| Reflection shell | *PLUS % possible obs: 75.8 % |
- Processing
Processing
| Software |
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.4→8 Å / σ(F): 2 / Stereochemistry target values: Engh & Huber Details: Initial coordinates for this structure were derived from the model of G. Cohen et al. (PDB entry 2GCH). Secondary structure elements and sequence data are identical to those of entry 2GCH ...Details: Initial coordinates for this structure were derived from the model of G. Cohen et al. (PDB entry 2GCH). Secondary structure elements and sequence data are identical to those of entry 2GCH except that the use of cryocrystallography techniques and the extended resolution of the current study permitted assignment of coordinates to residues 149 and 150, which are adjacent to a natural excission site. Initial phases were derived from the 2GCH model. A previous entry by Brady et al. (PDB entry 6gch) describes an identical complex determined by crystal structure analysis at lower resolution (2.1 Angstrom). More accurate determination of hydrogen bonding distances within the catalytic triad motivated the current study.
| |||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→8 Å
| |||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.5 / Classification: refinement | |||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.4 Å / Lowest resolution: 8 Å / σ(F): 2 / Rfactor obs: 0.175 | |||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||
| Refine LS restraints | *PLUS Type: x_angle_deg / Dev ideal: 1.4 |
