 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1gcz: MACROPHAGE MIGRATION INHIBITORY FACTOR (MIF) COMPLEXED WITH INHIBITOR. -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gcz | ||||||
|---|---|---|---|---|---|---|---|
| Title | MACROPHAGE MIGRATION INHIBITORY FACTOR (MIF) COMPLEXED WITH INHIBITOR. | ||||||
 Components Components | MACROPHAGE MIGRATION INHIBITORY FACTOR | ||||||
 Keywords Keywords | IMMUNE SYSTEM / PROTEIN-INHIBITOR COMPLEX / MIF / MACROPHAGE MIGRATION INHIBITORY FACTOR | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of prostaglandin secretion involved in immune response / positive regulation of myeloid leukocyte cytokine production involved in immune response / phenylpyruvate tautomerase / L-dopachrome isomerase / dopachrome isomerase activity / phenylpyruvate tautomerase activity / positive regulation of lipopolysaccharide-mediated signaling pathway / cytokine receptor binding / positive regulation of arachidonic acid secretion / negative regulation of myeloid cell apoptotic process ...positive regulation of prostaglandin secretion involved in immune response / positive regulation of myeloid leukocyte cytokine production involved in immune response / phenylpyruvate tautomerase / L-dopachrome isomerase / dopachrome isomerase activity / phenylpyruvate tautomerase activity / positive regulation of lipopolysaccharide-mediated signaling pathway / cytokine receptor binding / positive regulation of arachidonic acid secretion / negative regulation of myeloid cell apoptotic process / negative regulation of mature B cell apoptotic process / negative regulation of macrophage chemotaxis / positive regulation of chemokine (C-X-C motif) ligand 2 production / prostaglandin biosynthetic process / carboxylic acid metabolic process / regulation of macrophage activation / negative regulation of protein metabolic process / negative regulation of intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator / chemoattractant activity / protein homotrimerization / positive regulation of protein kinase A signaling / negative regulation of cellular senescence / negative regulation of DNA damage response, signal transduction by p53 class mediator / DNA damage response, signal transduction by p53 class mediator / positive regulation of B cell proliferation / positive regulation of phosphorylation / Gene and protein expression by JAK-STAT signaling after Interleukin-12 stimulation / negative regulation of cell migration / cytokine activity / positive regulation of cytokine production / Cell surface interactions at the vascular wall / positive regulation of fibroblast proliferation / cellular senescence / positive regulation of peptidyl-tyrosine phosphorylation / positive regulation of tumor necrosis factor production / positive regulation of peptidyl-serine phosphorylation / secretory granule lumen / protease binding / vesicle / ficolin-1-rich granule lumen / cell surface receptor signaling pathway / positive regulation of ERK1 and ERK2 cascade / inflammatory response / negative regulation of gene expression / innate immune response / positive regulation of cell population proliferation / Neutrophil degranulation / negative regulation of apoptotic process / cell surface / extracellular space / extracellular exosome / extracellular region / nucleoplasm / identical protein binding / plasma membrane / cytosol / cytoplasmSimilarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / molecular replacement / Resolution: 1.9 Å | ||||||
 Authors Authors | Katayama, N. / Kurihara, H. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2001 Title: Coumarin and chromen-4-one analogues as tautomerase inhibitors of macrophage migration inhibitory factor: discovery and X-ray crystallography. Authors: Orita, M. / Yamamoto, S. / Katayama, N. / Aoki, M. / Takayama, K. / Yamagiwa, Y. / Seki, N. / Suzuki, H. / Kurihara, H. / Sakashita, H. / Takeuchi, M. / Fujita, S. / Yamada, T. / Tanaka, A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gcz.cif.gz | 83.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gcz.ent.gz | 64.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1gcz.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gc/1gczftp://data.pdbj.org/pub/pdb/validation_reports/gc/1gcz | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1gd0SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / MIF Mass: 13426.209 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: PET22B / Species (production host): Escherichia coli / Production host:  Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: P14174 Escherichia coli BL21 (bacteria) / Strain (production host): BL21 / References: UniProt: P14174#2: Chemical | ChemComp-SO4 / Sulfate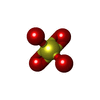  Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4 / Details: tautomerase inhibitor Mass: 96.063 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: SO4 / Details: tautomerase inhibitor#3: Chemical | Citric acid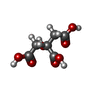  Mass: 192.124 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H8O7#4: Chemical |   Mass: 234.205 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H10O5 Mass: 234.205 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C12H10O5#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 137 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 50.15 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5 Details: ammonium sulfate, sodium citrate, pH 5.0, vapor diffusion/hanging drop, temperature 298.0K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: BL-6B / Wavelength: 1 / Beamline: BL-6B / Wavelength: 1 |
| Detector | Type: WEISSENBERG / Detector: DIFFRACTOMETER / Date: May 25, 1999 |
| Radiation | Monochromator: SI (1 1 1) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→20 Å / Num. obs: 31955 / % possible obs: 85.5 % / Redundancy: 3.1 % / Biso Wilson estimate: 16.9 Å2 / Rmerge(I) obs: 0.035 / Net I/σ(I): 8.6 |
| Reflection shell | Resolution: 1.8→1.85 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.489 / Num. unique all: 1939 / % possible all: 71.5 |
| Reflection | *PLUS Observed criterion σ(F): 2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: molecular replacement Starting model: 1GD0 Resolution: 1.9→20 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 Stereochemistry target values: R. A. Engh and R. Huber, Acta Cryst. Sect. A., 1991 Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.8 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→20 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.014 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file | Serial no: 1 / Param file: PARHCSDX.PRO / Topol file: TOPHCSDX.PRO | ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 98.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 20 Å / σ(F): 2 / % reflection Rfree: 9.8 % / Rfactor Rfree: 0.24 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 24.8 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.27 / % reflection Rfree: 10 % / Rfactor Rwork: 0.24 |
