 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fmu | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF NATIVE PROTEINASE A IN P3221 SPACE GROUP. | |||||||||
 Components Components | SACCHAROPEPSIN | |||||||||
 Keywords Keywords | HYDROLASE / Proteinase A | |||||||||
| Function / homology |  Function and homology informationsaccharopepsin / : / microautophagy / oligosaccharide binding / cytoplasm to vacuole targeting by the Cvt pathway / pexophagy / fungal-type vacuole / Neutrophil degranulation / proteolysis involved in protein catabolic process / macroautophagy ...saccharopepsin / : / microautophagy / oligosaccharide binding / cytoplasm to vacuole targeting by the Cvt pathway / pexophagy / fungal-type vacuole / Neutrophil degranulation / proteolysis involved in protein catabolic process / macroautophagy / autophagy / disordered domain specific binding / peptidase activity / aspartic-type endopeptidase activity / endoplasmic reticulum / protein-containing complex / mitochondrion Function and homology informationsaccharopepsin / : / microautophagy / oligosaccharide binding / cytoplasm to vacuole targeting by the Cvt pathway / pexophagy / fungal-type vacuole / Neutrophil degranulation / proteolysis involved in protein catabolic process / macroautophagy ...saccharopepsin / : / microautophagy / oligosaccharide binding / cytoplasm to vacuole targeting by the Cvt pathway / pexophagy / fungal-type vacuole / Neutrophil degranulation / proteolysis involved in protein catabolic process / macroautophagy / autophagy / disordered domain specific binding / peptidase activity / aspartic-type endopeptidase activity / endoplasmic reticulum / protein-containing complex / mitochondrionSimilarity search - Function | |||||||||
| Biological species |  Saccharomyces cerevisiae (brewer's yeast) Saccharomyces cerevisiae (brewer's yeast) | |||||||||
| Method | X-RAY DIFFRACTION / Resolution: 2.7 Å | |||||||||
 Authors Authors | Gustchina, A. / Li, M. / Phylip, L.H. / Lees, W.E. / Kay, J. / Wlodawer, A. | |||||||||
 Citation Citation | Journal: Biochem.Biophys.Res.Commun. / Year: 2002 Title: An unusual orientation for Tyr75 in the active site of the aspartic proteinase from Saccharomyces cerevisiae. Authors: Gustchina, A. / Li, M. / Phylip, L.H. / Lees, W.E. / Kay, J. / Wlodawer, A. #1: Journal: Nat.Struct.Biol. / Year: 2000Title: The aspartic proteinase from Saccharomyces cerevisiae folds its own inhibitor into a helix Authors: Li, M. / Phylip, L. / Lees, W. / Winther, J. / Dunn, B. / Wlodawer, A. / Kay, J. / Gustchina, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fmu.cif.gz | 78.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fmu.ent.gz | 62 KB | Display | PDB format |
| PDBx/mmJSON format | 1fmu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fm/1fmuftp://data.pdbj.org/pub/pdb/validation_reports/fm/1fmu | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | / PROTEINASE A Mass: 35774.551 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Saccharomyces cerevisiae (brewer's yeast) / References: UniProt: P07267, saccharopepsin | ||||||
|---|---|---|---|---|---|---|---|
| #2: Sugar | ChemComp-MAN / Mannose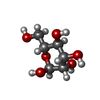  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 5 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 5Source method: isolated from a genetically manipulated source Formula: C6H12O6 #3: Sugar | N-Acetylglucosamine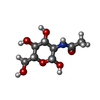  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #4: Sugar | ChemComp-NDG / | N-Acetylglucosamine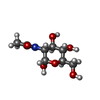  Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, alpha linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.14 Å3/Da / Density % sol: 60.8 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.6 Details: PEG 1500, Ammonium Sulfate, pH 5.6, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.6 | ||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Feb 6, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→30 Å / Num. all: 12565 / Num. obs: 12565 / % possible obs: 97.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 6.2 % / Biso Wilson estimate: 54.9 Å2 / Rmerge(I) obs: 0.064 / Net I/σ(I): 25.7 |
| Reflection shell | Resolution: 2.7→2.75 Å / Redundancy: 5.02 % / Rmerge(I) obs: 0.386 / Num. unique all: 613 / % possible all: 96.5 |
| Reflection | *PLUS Num. measured all: 78120 |
| Reflection shell | *PLUS % possible obs: 96.5 % / Mean I/σ(I) obs: 3.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 2.7→19.71 Å / Rfactor Rfree error: 0.012 / Data cutoff high absF: 3069059.21 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): -3 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 55.25 Å2 / ksol: 0.283 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 47.9 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→19.71 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.87 Å / Rfactor Rfree error: 0.037 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.7 Å / Rfactor Rfree: 0.2708 / Rfactor Rwork: 0.2071 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.32 |
