 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ekj: THE X-RAY CRYSTALLOGRAPHIC STRUCTURE OF BETA CARBONIC ANHYDRASE F... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ekj | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE X-RAY CRYSTALLOGRAPHIC STRUCTURE OF BETA CARBONIC ANHYDRASE FROM THE C3 DICOT PISUM SATIVUM | ||||||
 Components Components | BETA-CARBONIC ANHYDRASE | ||||||
 Keywords Keywords |  LYASE / ROSSMAN FOLD DOMAIN / STRAND EXCHANGE LYASE / ROSSMAN FOLD DOMAIN / STRAND EXCHANGE | ||||||
| Function / homology |  Function and homology information Function and homology informationcarbon utilization / chloroplast stroma / carbonic anhydrase / carbonate dehydratase activity / zinc ion bindingSimilarity search - Function | ||||||
| Biological species |   Pisum sativum (garden pea) Pisum sativum (garden pea) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.93 Å | ||||||
 Authors Authors | Kimber, M.S. / Pai, E.F. | ||||||
 Citation Citation | Journal: EMBO J. / Year: 2000 Title: The active site architecture of Pisum sativum beta-carbonic anhydrase is a mirror image of that of alpha-carbonic anhydrases. Authors: Kimber, M.S. / Pai, E.F. #1: Journal: To be PublishedTitle: Beta-Carbonic Anhydrase from Pisum Sativum: Crystallisation and Preliminary X-Ray Analysis Authors: Kimber, M.S. / Coleman, J.R. / Pai, E.F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ekj.cif.gz | 349.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ekj.ent.gz | 285.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1ekj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ek/1ekjftp://data.pdbj.org/pub/pdb/validation_reports/ek/1ekj | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 8 molecules ABCDEFGH
| #1: Protein | Mass: 23967.291 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Source: (natural) Pisum sativum (garden pea) / Cellular location: NUCLEAR ENCODED / Organ: LEAF / References: UniProt: P17067, carbonic anhydrase |
|---|
-Non-polymers , 8 types, 849 molecules 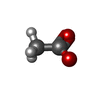



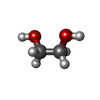

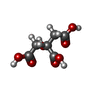

| #2: Chemical | ChemComp-ACT / Acetate Mass: 59.044 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: C2H3O2#3: Chemical | ChemComp-AZI / Azide Mass: 42.020 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: N3 Mass: 42.020 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: N3#4: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn#5: Chemical | ChemComp-CL / Chloride Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl#6: Chemical | ChemComp-EDO / Ethylene glycol Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H6O2#7: Chemical | ChemComp-CU / Copper Mass: 63.546 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cu Mass: 63.546 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Cu#8: Chemical | ChemComp-CIT / | Citric acid Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7#9: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 811 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.58 Å3/Da / Density % sol: 52.41 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 5.2 Details: 16% PEG 4000, 0.05 M DITHIOTHREITOL, 0.4 M AMMONIUM ACETATE, 0.1 M SODIUM CITRATE, pH 5.2, VAPOR DIFFUSION, HANGING DROP, temperature 277.0K | |||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 5 / Method: vapor diffusion | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Type: APS / Wavelength: 1 / Type: APS / Wavelength: 1 |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 1, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.93→40 Å / Num. obs: 138484 / % possible obs: 93.2 % / Observed criterion σ(I): -3 / Redundancy: 5.6 % / Biso Wilson estimate: 14.6 Å2 / Rmerge(I) obs: 0.067 / Net I/σ(I): 13 |
| Reflection shell | Highest resolution: 1.93 Å / Rmerge(I) obs: 0.277 / % possible all: 78.5 |
| Reflection | *PLUS Num. measured all: 779081 |
| Reflection shell | *PLUS % possible obs: 78.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.93→40 Å / Rfactor Rfree error: 0.007 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH AND HUBER Details: TORSION ANGLE ANNEALING REFINEMENT TO MAXIMIMUM LIKELIHOOD TARGETS, FOLLOWED BY INDIVIDUAL TEMPERATURE FACTOR REFINEMENT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 60.75 Å2 / ksol: 0.34 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 45.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.93→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.93→2.05 Å / Rfactor Rfree error: 0.025 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 0.5 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
