 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a5z | ||||||
|---|---|---|---|---|---|---|---|
| Title | LACTATE DEHYDROGENASE FROM THERMOTOGA MARITIMA (TMLDH) | ||||||
 Components Components | L-LACTATE DEHYDROGENASE Lactate dehydrogenase Lactate dehydrogenase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / GLYCOLYSIS / HYPERTHERMOPHILES / THERMOTOGA MARITIMA / PROTEIN STABILITY | ||||||
| Function / homology |  Function and homology informationL-lactate dehydrogenase / L-lactate dehydrogenase activity / glycolytic process / cytoplasm Function and homology informationL-lactate dehydrogenase / L-lactate dehydrogenase activity / glycolytic process / cytoplasmSimilarity search - Function | ||||||
| Biological species |   Thermotoga maritima (bacteria) Thermotoga maritima (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Auerbach, G. / Ostendorp, R. / Prade, L. / Korndoerfer, I. / Dams, T. / Huber, R. / Jaenicke, R. | ||||||
 Citation Citation | Journal: Structure / Year: 1998 Title: Lactate dehydrogenase from the hyperthermophilic bacterium thermotoga maritima: the crystal structure at 2.1 A resolution reveals strategies for intrinsic protein stabilization. Authors: Auerbach, G. / Ostendorp, R. / Prade, L. / Korndorfer, I. / Dams, T. / Huber, R. / Jaenicke, R. #1: Journal: Protein Sci. / Year: 1996Title: Extremely Thermostable L(+)-Lactate Dehydrogenase from Thermotoga Maritima: Cloning, Characterization, and Crystallization of the Recombinant Enzyme in its Tetrameric and Octameric State Authors: Ostendorp, R. / Auerbach, G. / Jaenicke, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a5z.cif.gz | 81.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a5z.ent.gz | 64 KB | Display | PDB format |
| PDBx/mmJSON format | 1a5z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a5/1a5zftp://data.pdbj.org/pub/pdb/validation_reports/a5/1a5z | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
-Protein / Sugars , 2 types, 3 molecules A
| #1: Protein | Lactate dehydrogenase Mass: 35026.387 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thermotoga maritima (bacteria) / Production host: Escherichia coli (E. coli) / References: UniProt: P16115, L-lactate dehydrogenase |
|---|---|
| #2: Sugar | Fructose 1,6-bisphosphate Type: D-saccharide, beta linking / Mass: 340.116 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 340.116 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C6H14O12P2 |
-Non-polymers , 4 types, 349 molecules 

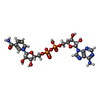

| #3: Chemical |  Mass: 112.411 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cd Mass: 112.411 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cd#4: Chemical | ChemComp-OXM / | Oxamic acid Mass: 89.050 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3NO3 Mass: 89.050 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3NO3#5: Chemical | ChemComp-NAD / | Nicotinamide adenine dinucleotide Mass: 663.425 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM#6: Water | ChemComp-HOH / | WaterMass: 18.015 Da / Num. of mol.: 344 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.8 Å3/Da / Density % sol: 68 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 / Details: pH 7.5 | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MPG/DESY, HAMBURG  / Beamline: BW6 / Wavelength: 1.5418 / Beamline: BW6 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 1, 1996 / Details: COLLIMATOR |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→20 Å / Num. obs: 30205 / % possible obs: 97.7 % / Redundancy: 3.7 % / Biso Wilson estimate: 28.3 Å2 / Rmerge(I) obs: 0.089 |
| Reflection | *PLUS % possible obs: 97.9 % / Num. measured all: 111661 |
| Reflection shell | *PLUS % possible obs: 96.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.1→8 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Cross valid method: THROUGHOUT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.4 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.8 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
